

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
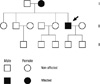
Familial hemiplegic migraine (FHM) is a rare form of migraine with aura that involves motor aura (hemiparesis) and an autosomal dominant classical migraine subtype. It can manifest with various neurological symptoms that are indistinguishable from stroke or seizure. The prevalence of hemiplegic migraine (HM) was reported as at least 0.002% for the sporadic form and at least 0.003% for the familial form in Denmark (1). In Korea, genetically confirmed HM has not been reported. Here, we present the first Korean family with FHM due to mutation in the CACNA1A gene.
CASE DESCRIPTION
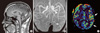
A 43-yr-old right-handed man presented to the Department of Emergency of Kangwon National University Hospital with suddenly noticed right hemianopia, dysphasia, and dizziness on March 11, 2010. When he arrived at the emergency room, global aphasia and vomiting developed. Subsequently, he became stuporous. Hemiparesis was not observed throughout the episode. Four and 5 yr before the admission, he had three episodes of right hemiparesis lasting a few hours. At those times, he was evaluated for a suspicion of transient ischemic attack, but without any risk factors for cerebral ischemia MRI showed cerebellar atrophy. Thus, he underwent genetic tests for mutations in the SCA1, 2, 3, 6, 7, 8, and DRPLA genes, which were all negative. He had occasional headaches, the nature of which could not be recalled because those were trivial. His family history revealed that his mother had suffered from headache and frequent transient hemiparesis, from her 30s to 50s (Fig. 1). Conventional MRI and diffusion-weighted imaging (DWI) performed when global aphasia developed showed only cerebellar atrophy (Fig. 2A). CT angiography performed during the coma, 3 hr after the current symptom onset, showed mild vasodilation of the intracranial blood vessels and increased vascularity in the left hemisphere (Fig. 2B). Perfusion-weighted imaging (PWI) showed elevated cerebral blood flow (CBF) (Fig. 2C). EEG revealed persistent low-voltage arrhythmic delta activity over the left hemisphere (Fig. 3). A cerebrospinal fluid study revealed no abnormalities.
A genetic study of FHM was performed using the samples from the patient and his mother, after obtaining their informed consents. Genomic DNA was extracted from the peripheral blood using an InstaGene Matrix Kit (Bio-Rad, CA, USA), according to the manufacturer's instructions. The entire coding region of the CACNA1A gene on Chr 19p13 was amplified via polymerase chain reaction using primer sets described previously and a Rotor-Gene 6000 thermal cycler. Direct DNA sequencing was performed using a BigDye® Terminator Cycle Sequencing Kit (Applied Biosystems, USA) on an ABI PRISM 3130 Genetic Analyzer (Applied Biosystems). The gene mutation analysis performed in the patient and his mother led to the identification of a heterozygous point mutation (1997C→T, T666M) in the exon 16 of the CACNA1A gene.
He recovered gradually over the following 4 days. Mild dysarthria and gait disturbance, which had existed before the event, were detected by repeated neurological examinations. Repeated PWI performed 10 days after recovery returned to normal. EEG gradually improved, but still showed a small amount of arrhythmic slowing in the left hemisphere, even after 10 days. We prescribed verapamil preventively and there have been no further attacks for the past 30 months.
DISCUSSION
There are three known types and corresponding loci of FHM. FHM1, which accounts for approximately 50% of FHM cases, is caused by mutations in the CACNA1A gene, which encodes the alpha-1 subunit of a high-voltage-gated P/Q type calcium channel (Cav2.1) (2). In familial and sporadic HM, more than 30 mutations have been identified in the CACNA1A gene. The mutation that causes the T666M substitution is the most common and accounts for 40% of unrelated FHM1 cases (3).
Previously, only one Korean family with symptoms that were suggestive of HM was reported, but without a genetic study (4). A few Korean families with different mutations in CACNA1A have been reported; however, they all exhibited episodic ataxia type 2 (5, 6).
Cav2.1 channels are located in the presynaptic terminals and in the somatodendritic membranes throughout the brain and play a role in controlling neurotransmitter release. The various mutations found in this calcium channel gene cause a wide spectrum of symptoms. FHM1 is associated with mostly missense mutations, whereas EA2 and SCA6 are associated with truncating mutations and expansion of a CAG repeat, respectively (7). Their effects on channel function were suggested to be a gain of function for the FHM1 mutation and a loss of function for the EA2 mutation (8). However, the phenotypes of FHM1, EA2, and SCA6 can coexist in a family member (7) and in single patient (9), suggesting that these allelic disorders form a broad disease spectrum.
Two thirds of FHM patients experience the cerebellar sign and about 30% of them exhibit cerebellar atrophy (10). This channel varies in type within different brain regions, and the Cav2.1 channels are mostly expressed in cerebellar Purkinje cells, which account for the progressive cerebellar sign in FHM1, EA2, and SCA6 (8). The T666M mutation is particularly associated with the cerebellar symptom and the high incidence of coma (2).
The episode of FHM presented here had distinctive features that did not fulfill the IHS diagnostic criteria (11), as the patient exhibited prolonged coma without hemiplegia and the absence of subsequent headache. A review of published cases showed that 38% of HM cases had no attack of hemiplegia (12). Coma affected up to a third of patients with FHM1 (3). One percent of FHM sufferers had no headache, with otherwise typical attacks (13). Aura might persist for weeks (14). Our patient showed different symptoms between attacks, but left-hemispheric symptoms were always present. Unilateral symptoms may switch between attacks, or the same side may always be affected (3).
Symptom variability among patients and attacks in a patient may be explained by genetic heterogeneity together with the probable presence of environmental and genetic modifying factors (2, 3). Symptom progression in the order of visual, sensory, motor, aphasic, and basilar disturbances is the common process (3), which can be explained by a cortical spreading depression (CSD) from the visual cortex (8). Symptom progression exceeding the vascular territory is a differentiating item from ischemia.
EEG during HM attacks usually shows attenuation or slow activities in the symptomatic hemisphere, which differentiates this condition from nonconvulsive seizures. The EEG abnormality outlasting the symptom in this patient could be the evidence of permanent sequelae of migraine, as well as cerebellar atrophy.
Cerebral hemodynamics in FHM has been reported anecdotally by angiography, PWI, or single photon emission computed tomography (SPECT). Most cases showed vasodilation or hyperperfusion during aura, which can differentiate this condition from ischemia (15-18). However, vasoconstriction was reported in a few cases (19, 20). Neither the severity of symptoms nor the time point of evaluation explained this discordance. No cases were identified genetically. We postulate that there can be heterogeneous pathophysiological abnormalities in these cases. In addition, many modifying factors may be involved in trigeminovascular activation secondary to electrophysiological dysfunction of neurons.
This is the first report of FHM in a Korean family and the only report of perfusion imaging in a genetically identified case. Because FHM is a rare disease, without a suspicion of HM, patients may be misdiagnosed as having transient ischemia, epilepsy, or even conversion disorder. Symptom progression exceeding the vascular territory from the initial visual symptom, absence of hypoperfusion, and electrical suppression differentiate HM from other episodic neurological disorders. FHM should be one of the differential diagnoses of episodic neurological dysfunction and cerebellar atrophy.

 XML Download
XML Download