

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Vitamin D is a steroidal hormone that is made available primarily through endogenous synthesis in the skin in response to ultraviolet light, after which it is metabolized in the liver and kidney (1). The effects of vitamin D are mediated by the binding of 1,25-dihydroxyvitamin D3 (1,25[OH]2D3) to a vitamin D receptor (VDR); a member of the steroid hormone receptor superfamily (2). The VDR is not only located in the tissues responsible for vitamin D metabolism and calcium homeostasis (kidney, intestine, and bone), but also in > 30 different tissues and cell lines. These tissues include the brain, breast, colon, heart, liver, prostate, pancreas, skin, stomach, and cells such as activated B- and T-lymphocytes and macrophages (3). Upon binding to the VDR in the nucleus, 1,25(OH)2D3 forms a heterodimeric complex with the retinoid x receptor (RXR) (4). The VDR-RXR complex binds to a hexameric binding motif in the promoter region of the vitamin D response element (VDRE) (5). The active metabolite of vitamin D3 (1α,25-dihydroxyvitamin D3, calcitriol) binds to the VDR and promotes interaction with VDREs in the promoter regions of target genes, resulting in transactivation of genes that induce cell cycle arrest, apoptosis, differentiation, antiangiogenesis responses, and inhibition of pro-growth/pro-survival signaling pathways (6, 7).
Studies using model systems of prostate, colon, and pancreatic cancer have shown that the administration of 1,25(OH)2D3 or vitamin D analogs produced significant anticancer effects (8-10). The effects of 1,25(OH)2D3 and its derivatives have been shown to function through the VDR for the regulation of proliferation, apoptosis, and angiogenesis (11-13). These data demonstrate the importance of 1,25(OH)2D3 as an antitumor agent. Unfortunately, the therapeutic applications of 1,25(OH)2D3 are limited because it is associated with hypercalcemia (14). Thus, new analogs of vitamin D that are potent but less calcemic have been synthesized and tested (15-17).
Paricalcitol (19-nor-1α-25-dihydroxyvitamin D2) is a synthetic analog of vitamin D that has been approved by the Food and Drug Administration for the clinical treatment of secondary hyperparathyroidism. This compound has very little calcemic activity, as demonstrated in randomized controlled clinical trials (18, 19). We designed this study to assess the antitumor and anti-inflammatory effects of paricalcitol in gastric cancer cells, and to evaluate the potential role of vitamin D in the treatment of peritoneal metastatic gastric cancer using in vivo models.
MATERIALS AND METHODS
Tumor cells and culture
Human gastric cancer cell line (MKN45) was obtained from the American Type Culture Collection (Manassas, VA, USA), and was grown as monolayers in RPMI 1640 medium (Invitrogen, Frederick, MD, USA) supplemented with 10% fetal bovine serum (FBS) (HyClone, Logan, UT, USA) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA, USA) at 37℃ in a humidified atmosphere containing 5% CO2.
Reagents
Paricalcitol (Abbott Laboratories, Chicago, IL, USA) was purchased as a 5 µg/mL stock solution dissolved in 30% propylene glycol and 20% alcohol. Paricalcitol was diluted in RPMI 1640 medium (Invitrogen) before use. Ethanol (≤ 0.001%) diluted in RPMI medium was used as a vehicle control.
Cell viability assay
Tumor cells were plated in 24-well tissue culture plates at 5 × 104 cells/well in RPMI-1640 medium containing 10% FBS and 1% penicillin-streptomycin sulfate. Following a 24-hr incubation at 37℃ in 5% CO2, the cells were treated with paricalcitol and incubated for 72 hr. The cells were pulsed with EZ-Cytox (Daeil Lab Service, Seoul, Korea) for 2 hr. Cellular absorption was then read at 450 nm with an ELISA reader (infinite M200 TECAN; Tecan Austria GmbH, Salzburg, Austria). The results are representative of at least 3 independent experiments.
Propidium iodide staining
Cells were plated on 6-well plates at a density of 1.5 × 105 cells/well overnight, followed by treatment with vehicle (control), or paricalcitol at 10 or 100 nM for 48 hr. Cells were trypsinized, washed with ice-cold phosphate buffered saline (PBS), and fixed with ice-cold 70% ethanol. Approximately 105 cells were resuspended in PBS containing propidium iodide (Sigma, St. Louis, MO, USA, 50 µg/mL) and RNase A (Sigma, 100 µg/mL) for 30 min in the dark and analyzed by flow cytometry using a FACS Calibur cell sorter (Becton Dickinson, San Jose, CA, USA) followed by quantification using CellQuest software (Becton Dickinson).
Western blot analysis
Aliquots containing 5 × 104 gastric cancer cells were seeded, incubated overnight, and treated with vehicle or paricalcitol at concentrations of 10 or 100 nM. Western blots were incubated overnight at 4℃ utilizing dilutions of primary antibodies against p21, p27, CDK2, CDK4, CDK6, Cyclin D1, Cyclin E, Bax, Lamin-A (Santa Cruz Biotech, Santa Cruz, CA, USA), Bcl-XL, Caspase 3, cleaved caspase 3, COX-2, STAT3, phospho-STAT3 (Cell Signaling, Danvers, MA, USA), NF-kappa B p65 (Imgenex, San Diego, CA, USA), and actin (Abcam, Cambridge, GBR). The cells were treated with paricalcitol and harvested for immunoblot analysis 48 hr after treatment with the inhibitor. The cellular proteins were separated, transferred, and immunoblotted. Immunoreactive bands were visualized using a secondary horseradish-peroxidase-conjugated anti-rabbit or anti-mouse antibody and the Western blot Detection Kit, chemiluminescent system (Millipore, Billerica, MA, USA). The blots were reprobed with anti-actin antibodies (Santa Cruz Biotech) as a loading control.
Fluorescence microscopy
Apoptotic cells were quantified by fluorescence microscopy using the Annexin V Apoptosis Detection Kit (BD Pharmingen, San Diego, CA, USA). The gastric cells (5 × 104/mL) were seeded in an 6-well plates, followed by treatment with paricalcitol 10 nM or 100 nM and stained with 5 µL of annexin V-Allophycocyanin (APC) at room temperature for 15 min in the dark. After staining, the cancer cells were washed with binding buffer, placed on a glass slide and covered with a glass cover slip. The stained cells were observed under a fluorescence confocal microscope FLUOVIEW FV1000 (Olympus, Tokyo, Japan) using filter sets for APC and 4'6-diamidino-2-phenylindole (DAPI).
Animal studies
BALB/c mice (5-7 weeks of age) were obtained from Jungang Lab Animal Inc. (Seoul, Korea). The Chonnam National University Medical School Research Institutional Animal Care and Use Committee approved the experimental protocol.
Intraperitoneal xenografts were established by injecting MKN 45 cells (1 × 107 cells per mouse) into the abdominal cavity of BALB/c mice. The mice were blindly and randomly assigned to either the experimental group or the control group. Treatment started 1 week after the MKN 45 cells were injected and continued for 4 weeks. The 5 control mice received vehicle (100 µL of PBS containing 20% ethanol per day, subcutaneously, 3 days per week on Monday, Wednesday, and Friday) only, and the 5 experimental mice received paricalcitol (100 ng in 100 µL of PBS containing 20% ethanol per day, subcutaneously, 3 days per week on Monday, Wednesday, and Friday). After 4 weeks, the mice were examined by micro-positron emission tomography (microPET) and blood was collected to measure the level of serum calcium. All mice were sacrificed, weighed, and the mean number of tumor nodules in the peritoneum of each mouse was recorded.
MicroPET protocol
All mice were examined in a fasted status. A solution containing 0.3 mCi 18F-fluorodeoxyglucose (18F-FDG) was injected into the tail vein of each mouse, and anesthesia was performed with 3% isoflurane (in combination with O2 at a flow rate of 2 L/min) 40 min later. PET data acquisition was performed using a Siemens Inveon system (Berlin, Germany) 1 hr after anesthesia when the mice were totally unconscious, and the procedure required 20 min for each mouse. Anesthesia was continued during the scanning process with the same flow rate through a face-mask designed for small animals. All microPET images were studied on a high-resolution computer screen using PMOD software (version 3.204) (PMOD technologies, Zurich, Switzerland). Both visual and semiquantitative analyses were performed for each mouse. For visual analysis, we compared the intra-abdominal color intensity produced by 18F-FDG uptake in each mouse from the 2 groups. For semiquantitative analysis, 3D round regions of interests (ROI) were carefully placed over the intra-abdomen area on microPET images of each mouse. The 18F-FDG uptake was recorded as a standardized uptake value (SUV).
RESULTS
Antiproliferative effects of paricalcitol in gastric cancer cells
We examined the cytotoxicity of paricalcitol in MKN45 cell line. As expected, paricalcitol inhibited the growth of gastric cancer cells in a dose-dependent manner (Fig. 1). To determine the effect of paricalcitol on cell cycle progression, gastric cancer cells were treated with 10 nM or 100 nM paricalcitol for 48 hr. This treatment promoted the accumulation of cells in the G0/G1 phase of the cell cycle, with a subsequent decrease in the number of cells in the S and G2/M phases. The percentage of cells in the G0/G1 phase increased by up to 30% and the percentage of cells in the S phase decreased by up to 70% (Fig. 2), indicating that aricalcitol effectively inhibits cell cycle progression.
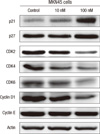
Effects of paricalcitol on expression of cell cycle regulators
Activation of the VDR results in several downstream effects, such as cell cycle arrest and apoptosis. To explore the mechanism by which vitamin D analogs induce G0/G1 cell cycle arrest in gastric cancer cells, the effect of paricalcitol on the expression of G1 phase cell cycle regulators, such as p21, p27, and CDK2, was determined. Gastric cancer cells were incubated with 10 or 100 nM paricalcitol for 48 hr and the levels of cell cycle regulator proteins were measured. The level of p21 protein was 2.9-fold higher in cells treated with 100 nM paricalcitol than in the control cells. The expression of P27 protein was also increased, though not in a statistically significant manner. The expression of CDK2, CDK4, CDK6, and Cyclin D1 protein was significantly decreased after treatment with paricalcitol. However, the expression of Cyclin E was not significantly changed (Fig. 3). The increased expression of cyclin-dependent kinase inhibitors following paricalcitol treatment may account for the observed cell cycle arrest.
Effects of paricalcitol on apoptosis in gastric cancer cells
The effect of paricalcitol on apoptosis was examined in gastric cancer cells. To investigate the involvement of apoptosis, protein expression levels of Bax, Bcl-XL, Caspase 3, and cleaved-Caspase 3 in cultured gastric cells were studied after treatment with paricalcitol. As shown in Fig. 4A, the levels of pro-apoptotic Bax and Caspase 3 (17 kDa) were significantly increased, whereas the level of anti-apoptotic Bcl-XL was decreased following paricalcitol treatment.
To determine the effect of paricalcitol on apoptosis, gastric cancer cells were treated with 10 or 100 nM paricalcitol for 48 hr, because these concentrations significantly increased the proportion of apoptotic cells. Apoptosis-associated changes in the cells were examined by fluorescence microscopy. As shown in Fig. 4B, more apoptotic events were observed in paricalcitol treated cells than in control cells.
Effects of paricalcitol on inflammation-dependent tumor promotion
NF-κB signaling is highly active in the epithelial cells of inflamed tissues. NF-κB is an important regulator of pro-inflammatory signals, programmed cell death and drug resistance, and strongly induces COX-2 gene expression. We tested whether paricalcitol could reduce inflammatory gene expression, such as those of COX-2 and nuclear NF-κB. Paricalcitol significantly suppressed expression of the inflammatory cytokine, COX-2 (Fig. 5). To assess whether this change was caused by reduced transcription of the COX-2 gene mediated by inactivation of the redox-sensitive transcription factor NF-κB, we performed Western blot analysis of NF-κB p65 in the nuclear fraction of whole cell extracts. Levels of nuclear NF-κB p65 were, indeed, significantly attenuated, suggesting that paricalcitol suppresses the expression of inflammatory mediators by reducing the level of NF-κB DNA in the nucleus.
STAT3 signaling is a major intrinsic pathway for cancer inflammation because it is frequently activated in malignant cells and induces a large number of genes that are crucial for inflammation. Our results indicate that paricalcitol significantly reduced the levels of phosphorylated STAT3 (Fig. 5).
Effects of paricalcitol in peritoneal metastases model
In visual analysis, an intense intra-abdominal uptake of 18F-FDG was found as demonstrated by the microPET images (Fig. 6A). Paricalcitol treatment produced a decreased uptake in mice. In a semiquantitative analysis, 18F-FDG uptake was significantly lower in the paricalcitol group compared to control group (SUV; control group 13.2 ± 5.3 vs paricalcitol group 4.5 ± 3.0, P = 0.005) (Fig. 6B).
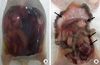
Bloody ascites and mesenteric nodules were recognized in the peritoneal metastases model (Fig. 7). Intraperitoneal tumor volume was significantly lower in paricalcitol treated mice. While the number of peritoneal nodules in paricalcitol treated mice tended to be lower than the number in vehicle treated mice, there were no statistically significant differences between the two groups in body weight, volume of ascites, or the number of peritoneal nodules. Serum calcium levels were 10.7 mg/dL in control mice and 9.9 mg/dL in paricalcitol-treated mice; both within the normal range (Table 1).
DISCUSSION
Vitamin D analogs have been examined pre-clinically for their therapeutic efficacy in preventing and treating cancer. The effects of vitamin D analogs have been shown to function through the VDR for the regulation of proliferation, apoptosis, and angiogenesis (9, 20, 21). We observed that the vitamin D analog, paricalcitol, inhibited proliferation of gastric cancer cell lines in vitro by modulating cell cycle progression, apoptosis, and inflammatory mediators.
The antiproliferative activity of paricalcitol was accompanied by cell cycle arrest and changes in the expression of p21 and CDK2. Paricalcitol caused gastric cancer cells to arrest in the G0/G1 phase, and induced the expression of p21. The CDKIs may play a role in the antiproliferative effects of paricalcitol by reducing the ability of the tumor cells to enter S phase (22). This study demonstrated that paricalcitol decreased CDK2 expression. CDK2 is a positive regulator of cell cycle progression (23); therefore, its inhibition by paricalcitol could contribute to the antitumor effects of this vitamin D analog.
Calcitriol exerts antitumor effects by reducing the expression of key mediators of apoptosis, such as anti-apoptotic, pro-survival proteins Bcl-2 and Bcl-XL, and by inducing the expression of pro-apoptotic proteins, such as Bax, Bak, and Bad (11). It has been reported that calcitriol downregulates Bcl-2 expression in MCF-7 breast tumor cells and HL-60 leukemia cells and upregulates Bax and Bak expression in prostate cancer, colorectal adenoma, and carcinoma cells (11). In addition to regulating the expression of the Bcl-2 family, calcitriol might also directly activate caspase effector molecules, although it is unclear whether calcitriol-induced apoptosis is caspase-dependent. In this study, paricalcitol significantly induced apoptotic cell death in gastric cancer cells. Furthermore, the molecular response of gastric cancer cells to the paricalcitol was similar to the effect of calcitriol in other cancer cells.
STAT3 and NF-κB have crucial and integrated roles in the inflammatory responses that promote cancer development and growth (24, 25). STAT3 also participates in oncogenesis by upregulating genes encoding the antiapoptotic proteins Bcl-XL and survivin, as well as the cell cycle proteins cyclin D1 and c-Myc, and inducers of angiogenesis (26). In the present study, paricalcitol treatment strongly inhibited STAT3 phosphorylation. However, we did not evaluate the expressions of STAT3 regulated genes such as cyclin D1, c-Myc and survivin in this study. Later we need to confirm regulation of STAT3-mediated inflammation. It has been shown that NF-κB is involved in modulating the expression of several pro-inflammatory genes, including COX-2 (27). Activation of NF-κB leads to the upregulation of the downstream COX-2 gene, which has been linked to cancer. In the present study, we found that paricalcitol reduced NF-κB and concomitantly inhibited the expression of COX-2. Thus, paricalcitol could partly potentiate anti-inflammatory activity by modulating NF-κB and its downstream genes.
Peritoneal metastasis is the most common type of metastasis in advanced gastric cancer, and it has a strong negative effect on patient prognosis (28). In this study, paricalcitol inhibited the in vivo growth of peritoneal gastric cancer in mice, as determined by microPET. The tumors on the mesentery in the control group were significantly larger than those in the paricalcitol group. However, there were no significant differences in the number of peritoneal nodules, bloody ascites, or the body weights. Although the number of peritoneal nodules did not significantly differ between the control and the paricalcitol groups, there may have been fewer nodules on the mesentery in the paricalcitol group (Control group 3.2 ± 1.5 vs paricalcitol group 1.8 ± 0.8).
In conclusion, paricalcitol has anticancer activity in gastric cancer cells by regulation of the cell cycle, apoptosis, and inflammation.






 XML Download
XML Download