

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Fabry's disease, sometimes referred to as Anderson-Fabry disease, is a rare X-linked disorder caused by deficient activity of the lysosomal enzyme, α-galactosidase A (GLA), and the progressive accumulation of glycosphingolipid globotriaosylceramide (Gb3) in various organ systems (1, 2). Fabry's disease is characterized clinically by chronic pain and acroparesthesia, gastrointestinal disturbances, characteristic skin lesions (angiokeratoma), progressive renal impairment, cardiomyopathy, and stroke. The causative gene, the so-called GLA gene, is located on the X-chromosome at q22.1, and more than 150 mutations of this gene have been detected (3). On the other hand, congenital agammaglobulinemia is a rare genetic disorder characterized by a dramatically reduction or absence of mature B lymphocytes in peripheral blood (usually < 2% of total lymphocytes), caused by differentiation arrest affecting the transition of B-cell progenitors into mature B lymphocytes (4). The Bruton tyrosine kinase (Btk) gene, located in the Xq21.3-q22 region at the long arm of chromosome X, has been shown to be the major causative gene of congenital agammaglobulinemia (5).
Both Fabry's disease and congenital agammaglobulinemia are rare genetic disorders, and to our knowledge, no case of Fabry's disease and congenital agammaglobulinemia co-occurrence has previously been reported. Here, we describe the case of a Korean man with both syndromes.
CASE DESCRIPTION
A 23-yr-old Korean man presented with multiple tiny red to purple papules on his back and scrotum. Although he first noticed the lesions 5 yr previously, he has received no treatment. Prior to presentation, he had noticed that the numbers were increasing, and thus, he visited our dermatology department for evaluation and work-up on 13 October 2008.
He had been on immunoglobulin replacement (IVIG 300-400 mg/kg monthly) therapy since he had received a diagnosis of congenital agammaglobulinemia at 5 yr. Furthermore, he had a history of recurrent respiratory infections, acroparesthesia, and hypohidrosis from childhood, and had presented with persistent and progressive proteinuria from 14 yr.
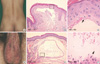
The patient was born to nonconsanguineous parents and there was no familial history. A physical examination revealed multiple 2-4 mm sized, red to purple papules on his back and scrotum (Fig. 1A, B). Neurologic examination results were normal aside except for patchy temperature sensation impairment on the lower legs and feet.
Skin biopsies of the angiomatous lesions on his back and scrotum, commonly showed dilated capillaries in the papillary dermis and sometimes vacuoles within endothelial cells (Fig. 1C, D). Complete blood count, electrolytes, complements, and liver, kidney, and thyroid function tests were non-specific. However, an immunoglobulin series revealed that almost all immunoglobulins, except IgD and IgG, were much reduced; immunoglobulin replacement therapy had probably resulted a near normal IgG level (802 mg/dL, ref.: 880-1,800 mg/dL).
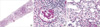
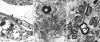
Furthermore, the B cell percentage was markedly lower at < 0.1% by lymphocyte analysis, and proteinuria (random urine P/Cr = 3.94 g/g, microalbumin > 500 µg/mL, 24-hr urine protein excretion = 3.192 g) was still evident. Electrocardiography findings suggested left ventricular hypertrophy. Renal ultrasonographic findings were normal, but a histopathologic examination of a kidney specimen showed global and segmental sclerosis in glomeruli with moderate fibrosis in interstitia, and diffuse foamy changes within the cytoplasm of podocytes and tubular epithelial cells (Fig. 2). Electron microscopic findings showed numerous electron dense lamellar inclusion bodies (Gibra bodies) in the cytoplasm of skin fibroblasts and renal podocytes (Fig. 3). On ophthalmologic evaluation revealed the presence of fine, whorl-like superficial corneal opacities, and vertex dystrophy (Fig. 4).
A GLA enzyme activity assay showed 105 nmole/hr/µg (normal control: 2,744), representing a reduction to only 4% of normal. We received a written consent from his parents and performed the genetic analysis. DNA PCR-sequence analysis identified an intronic variation on the splicing acceptor site in the GLA gene (Fig. 5A), and direct sequencing studies of peripheral blood DNA identified the presence of a GLA gene mutation (Fig. 5B) but revealed no genetic abnormality at the Btk gene locus, which is shown by 85% of patients with congenital agammaglobulinemia. As a result of these clinical and laboratory findings, he has diagnosed as having both Fabry's disease and non-Bruton type congenital agammaglobulinemia. He is being treated with intravenous gammaglobulin (400 mg/kg) monthly for the congenital agammaglobulinemia and with oral ACE inhibitors for proteinuria. GLA enzyme replacement therapy is planned to treat Fabry's disease.
DISCUSSION
Fabry's disease is an X-linked metabolic disorder that was initially described in 1898. The incidence of Fabry's disease has been estimated at one per 40,000 to 117,000 worldwide (6). Deficiency of GLA leads to the storage of neutral glycosphingolipids, particularly of Gb3, in many tissues and cell types. The progressive accumulation of these molecules eventually leads to cellular dysfunction, and possibly to inflammation and/or fibrosis. These processes lead to organ dysfunction and clinical evidence of Fabry's disease. The mechanism of tissue damage is believed to be at least partly due to poor perfusion caused by accumulations in the vascular endothelium, particularly in kidneys, heart, the nervous system, and skin either alone or in combination with deposits in other cell types (2).
The progression of clinical symptoms in Fabry's disease can be considered conceptually to follow patient age. Early symptoms in children include acroparesthesia, hypohidrosis, and gastrointestinal symptoms, such as, nausea, abdominal pain, and postprandial diarrhea (7, 8). However, from age 20 yr, these symptoms tend to progress and proteinuria appear. Renal failure is usually encountered in most men with the disease with concomitant progression in other affected organ systems, which leads to life-threatening cardiac and cerebrovascular manifestations and substantial morbidity (8).
The GLA gene, the causative gene of Fabry's disease, is located in the Xq22.1 region. It is about 12 kb long and contains 7 exons that encode a precursor protein of 429 amino acids. The basic molecular defects in Fabry disease include partial deletion, duplication, and point mutation of the GLA gene (9). Currently, more than 400 mutations have been detected in this gene.
Congenital agammaglobulinemia is a rare humoral immunodeficiency disease. The prevalence of congenital agammaglobulinemia has been estimated at one per 200,000 worldwide (10). Affected individuals have markedly reduced levels of all major classes of immunoglobulins in serum, and increased susceptibility to recurrent bacterial infections (11-13). The most common symptoms of congenital agammaglobulinemia are sinusitis, bronchitis, otitis, and pneumonia, and symptoms usually appear in infancy or early childhood after maternal immunoglobulins have been lost. Approximately 85% of patients with congenital agammaglobulinaemia have the X-linked form due to mutations in the Btk gene (12). The remaining 15% of cases, which clinically are inseparable from X-linked agammaglobulinemia cases, have an autosomal recessive or idiopathic form.
The Btk gene, the most common causative gene of congenital agammaglobulinemia, is located in the Xq21.3-q22 region, and was first identified in 1993 (13). It spans 37.5 kb of genomic DNA, consists of 19 exons (14), and encodes a 77 kDa cytoplasmic protein tyrosine kinase containing five domains, that is, the N-terminal pleckstrin homology (PH), the proline-rich Tec homology (TH), the Src homology 3 (SH3), the Src homology 2 (SH2), and the catalytic tyrosine kinase (TK) domains (15). Btk is expressed by all hematopoietic lineages, excepting T lymphocytes and plasma cells, and plays an essential role in the development of B cells. Our ability to identify Btk gene mutations probably explains why X-linked agammaglobulinemia is the most commonly diagnosed type of congenital agammaglobulinemia, and to date more than 600 unique Btk gene mutations have been recorded in the BTKbase (a designated international mutation database) (10).
The described case had symptoms and signs typical of both Fabry's disease and congenital agammaglobulinemia, respectively. Several disease clusters are located around loci in the Xq22 region, such as, those of Alport syndrome, Fabry's disease, and X-linked agammaglobulinemia. In 1995, Oeltjen et al. (16) reported that the Btk gene and the GLA gene are localized in the same 50 to 70 kb region. However, the relationships between genes in this region have not established despite their contiguities. A genetic study was undertaken to detect GLA and Btk genes abnormalities in present case to investigate whether the co-occurrence of the two syndromes was incidental or inevitable. However, we were unable to find any abnormality in the Btk gene or any specific genetic relationship between the two disorders, and thus, it appears that their co-occurrence in our patient was coincidental.
Here, we report the first case of Fabry's disease with congenital agammaglobulinemia in 23-yr-old man. Although we were unable to find any abnormality in the Btk gene or any specific genetic relationship between the two disorders, this case provided a rare and interesting opportunity to consider these two syndromes on a genetic basis. It is suggested that more genetic studies should be performed to identify the association between Fabry's disease and congenital agammaglobulinemia.


 XML Download
XML Download