

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatic ischemia reperfusion (I/R) injury is a pathophysiologic process whereby hypoxic organ damage is accentuated following return of blood flow and oxygen delivery (1-3). Transient episodes of tissue ischemia are encountered during solid organ transplantation, trauma, hypovolemic shock and elective liver resection, when inflow occlusion or total vascular exclusion is used to minimize blood loss. In these situations, after a period of ischemia, the liver can be significantly injured upon its reperfusion. If the injury is severely uncontrolled, this can lead to liver failure, systemic inflammatory response syndrome and multiple organ failure which are associated with high rates of morbidity and mortality (4).
I/R injury is characterized by an excessive inflammatory response after reperfusion, which exert a crucial role in the pathophysiology of postischemic hepatic failure (5). The injury occurs in a biphasic pattern: The early pattern is characterized by hepatic injury occurring within 1-6 hr after reperfusion, associated with Kupffer cell activation and up-regulation of reactive oxygen species, the inducible nitric oxide synthase and the pro-inflammatory cytokines (6, 7). Subsequently, the injury is aggravated by a massive neutrophil infiltration, peaking 9-24 hr following reperfusion. The neutrophils are recruited into the liver vasculature, which is activated by various cellular adhesion molecules. These neutrophils cause hepatocyte necrosis through the release of cytotoxic proteases and oxygen-derived radicals.
The neutrophil recruitment during inflammation is classically attributed to a multi-step cascade involving initial tethering and rolling along the vessel wall, followed by firm adhesion to the vascular endothelium and emigration out of the vasculature. This classical paradigm is well characterized for a number of organ microvasculatures including mesentery, peritoneum, skeletal muscle, and skin (8). Within the post-capillary vessels of these tissues, the classical paradigm of neutrophil recruitment involve selectin mediated tethering and rolling, followed by integrin mediated firm adhesion (9). The liver has two principal vascular beds for neutrophil recruitment during reperfusion: sinusoids and postsinusoidal venules. The evidence for transmigration from postsinusoidal venules is limited. In contrast, sinusoids were identified as the dominant sites for neutrophil extravasation (10). The sinusoidal endothelium is the unique structure of the liver, which is discontinuous, fenestrated, and lacking basal lamina and tight junction. And then in these narrow capillaries, the recruitment of slow-moving leukocytes is selectin independent, and the rolling process is likely to be unnecessary. In short, the liver presents an apparent exception to this classical paradigm of neutrophil recruitment in three ways: 1) in addition to the postsinusoidal venules, neutrophils also adhere to the endothelium of capillaries called sinusoids, 2) within the sinusoids, neutrophils appear not to roll for a significant distance, but rather tether and immediately adhere and 3) the adhesion in the sinusoids is shown to be completely independant of selectin (11, 12).
Some clinical trials of anti-adhesion therapy in an attempt to reduce injury associated with I/R injury failed to show a significant benefit, despite very strong preclinical data (13). Recently, several adhesion molecules, including VAP-1, CD44, CD43, and Fcγ RIII for lymphocyte adhesion in sinusoidal endothelium have been proposed (14). Therefore, we examined the role of CD44 in liver reperfusion injury, at various lengths of reperfusion time. In current study, we sought to test whether or not hepatic I/R injury would be attenuated in mice treated with anti-CD44 antibody before I/R insult, at time points most consistent with the neutrophil-mediated phage of liver injury.
MATERIALS AND METHODS
Animals
Male wild-type (C57BL/6) mice (8-12 week old) were purchased from the Jackson Laboratory. All animals were maintained in a laminar-flow, specific pathogen-free atmosphere at the Eulji University School of Medicine. The protocol for animal experiment in this article was previously approved by the Institutional Animal Care and Use Committee of the Eulji University School of Medicine (EIACUC 09-01). All subsequent animal experiment adhered to the "Regulation for Animal Expermentation" of the University.
Liver ischemia
A nonlethal model of segmental (70%) hepatic warm ischemia was used (15). The I/R protocol was initiated with the abdominal wall being clipped of hair and cleansed with betadine. Under sodium pentobarbital (40 mg/kg, intraperitonal) and methoxyflurane (inhalation) anesthesia, a midline laparotomy was performed. With the use of an operating microscope, the liver hilum was dissected free of surrounding tissue. All structures in the portal triad (hepatic artery, portal vein, and bile duct) to the left and median liver lobes were occluded with a microvascular clamp (Fine Science Tools) for 60 min, and reperfusion was initiated by removal of the clamp. This method of segmental hepatic ischemia prevents mesenteric venous congestion by permitting portal decompression through the right and caudate lobes. We have previously conducted a time course experiment to determine the optimal ischemia time period for the induction of hepatic injury. Less than 60 min of ischemia produced only minimal plasma transaminase elevations, whereas over 75 min of ischemia was poorly tolerated with gross evidence of poor reperfusion of the ischemic lobes. A reproducible level of hepatic damage was observed using 60 min of ischemia and was thus used for our study. After application of the clamp, the abdomen was covered with a sterile plastic wrap to minimize evaporative loss. Throughout the ischemic interval, evidence of ischemia was confirmed by visualizing the pale blanching of the ischemic lobes. The clamp was then removed and gross evidence of reperfusion based on immediate color change was assured before closing the abdomen with a continuous 4-0 diameter polypropylene suture. Either the absence of ischemic color changes or the lack of response to reperfusion was a criterion for immediate sacrifice and exclusion from further analysis. Temperature was monitored by rectal temperature probe and was maintained at 37℃ by means of a warming pad and heat lamp. At the end of the observation period following reperfusion, the mice were anesthetized with inhaled methoxyflurane and were sacrificed by exsanguination. Sham animals underwent anesthesia, laparotomy, and exposure of the portal triad without hepatic ischemia. Animals were sacrificed at predetermined time points (6-72 hr) after reperfusion to obtain serum and liver samples. Antibody-treated mice received 16 hr before surgery a single intraperitoneal injection of 100 µg of anti-CD44 mAb (16) (clone IM 7, purified rat anti-mouse CD44; BD Pharmingen, San Diego, CA, USA) that interacts with the HA-binding site of CD44 and may induce shedding of CD44 or control IgG (normal rat IgG; Santa Cruz, CA, USA).
Peripheral blood and tissue procurement
Blood samples were collected from the right ventricle via median sternotomy in a sterile heparinized syringe containing 50 µL of heparin (100 USP units/mL). The blood samples were centrifuged and plasma were collected and stored at -80℃ until further use. Portions of the ischemic and non-ischemic liver lobes were fixed in buffered 10% formalin, embedded in paraffin, and used for hematoxylin and eosin (H&E) staining and immunohistochemical study. Other portions of ischemic and non-ischemic liver lobes were snap frozen in liquid nitrogen and stored at -80℃, until use for western blotting analysis.
Liver damage assessment
To assess hepatic function and cellular injury following liver ischemia, serum alanine aminotransferase (ALT) levels were measured using the Opera Clinical Chemistry System (Bayer, Munich, Germany).
Plasma cytokine concentrations
Plasma TNF-α, IL-6, and MCP-1 levels were determined in a 96-well Nunc-Immuno microplate (VWR Scientific, Chicago, IL, USA), using a sandwich enzyme-linked immunosorbent assay (ELISA). The capture antibody was a polyclonal anti-mouse TNF-α, IL-6, or MCP-1 specific goat IgG (R&D Systems, Minneapolis, MN, USA) and the detection antibody was a biotinylated polyclonal anti-mouse TNF-α, IL-6, or MCP-1 specific goat IgG (R&D Systems). All plasma samples were tested in duplicate. The minimal detectable protein concentration was 20 pg/mL.
Histopathological study
Formalin-fixed liver samples were embedded in paraffin and cut to 5 µm thick sections. Tissues were stained with hematoxylin-eosin, and slides were assessed for inflammation and tissue injury. The percent necrotic area was estimated by random evaluation of lower power fields (× 40) of each hematoxylin-eosin-stained section. A pathologist, blinded to the experimental procedure of the mice, examined the histopathology of the hepatic tissue sections.
Immunohistochemical staining
Tissue sections in 5 µm thickness were prepared from paraffin-embedded tissue blocks. Tissue sections were deparaffinized in xylene and rehydrated in a series of ethanol. Microwaving antigen retrieval was performed in 0.01 M citrate buffer, pH 6.0, for 10 min. Then, tissue sections were placed in 0.3% H2O2/methanol for 15 min to inactivate endogenous peroxidase. After washing in PBS, tissue sections were incubated in 10% normal goat serum (Chemicon International, Temecula, CA, USA) for 30 min at room temperature to block nonspecific binding of the primary antibody. Then, tissue sections were exposed to diluted primary antibody for overnight at 4℃. Proper dilutions of primary antibodies were selected from preliminary studies. We used dilutions of 1:200 for CD44 (monoclonal rat anti-mouse CD44; BD Pharmingen, Franklin Lakes, NJ, USA) and 1:25 for neutrophil (monoclonal rat anti-neutrophil; Abcam, Cambridge, MA, USA). After washing in PBS to remove excess primary antibody, tissue sections were then incubated with biotinylated goat anti-rat IgG (SC-2041, Santa Cruz, Santa Cruz, CA, USA) at 1:100 dilution for 1 hr at room temperature. Unbound secondary antibody was removed with PBS washing, and avidin-biotin peroxidase (Vector Laboratories, Burlingame, CA, USA) was placed on tissue sections for 30 min at room temperature. The positive immune-reactivity on tissue sections was detected by treatment with a mixture of 3,3'-diaminobenzidine (Sigma, St. Louis, MO, USA), 0.05 M Tris-HCl buffer, and 5% hydrogen peroxide. Then, the tissue sections were counterstained with hematoxylin, followed by dehydration in a series of ethanol. Immunostaining was examined and evaluated under light microscopy. For negative control, normal goat serum at the same dilutions in the place of primary antibody was treated on tissue sections.
SDS-PAGE and western blot analysis
After protein concentration was determined using the BCA protein assay, equal volumes of elute were fractioned by SDS-PAGE and transferred to nitrocellulose membranes for 2 hr at 250 mA in the presence of transfer buffer. The transferred membrane was blocked for 1 hr at room temperature with 5% milk in TBS-T buffer and incubated with purified rat anti-mouse CD44 mAb (KM114; BD Pharmingen), rat anti-mouse neutrophil mAb (clone 7/4; Abcam) respectively for 12 hr at 4℃. Blots were incubated at room temperature for 1 hr with a horseradish peroxidase-conjugated secondary antibody against the primary antibody. After extensive washing with TBS-T buffer, membranes were developed with the Super Signal West Pico chemiluminescent kit (Pierce Chemical Co., IL, USA) and exposed to film.
RESULTS
Demonstration of hepatocellular injury by changes in reperfusion time
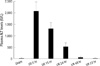
Hepatic I/R caused significant hepatocellular damage as demonstrated by plasma ALT levels. The plasma ALT levels of wild-type mice after 60 min. of ischemia followed by serial time interval of reperfusion were measured. The plasma ALT level was maximally elevated after 6 h of reperfusion following ischemic insult, which was decreased to level of sham operation after 72 hr (Fig. 1).
Demonstration of inflammatory cytokine levels by changes in reperfusion time
In order to determine whether plasma cytokine/chemokine levels correlated with tissue injury, the plasma cytokine/chemokine levels were measured using an ELISA. The plasma levels of all cytokines/chemokine (i.e., TNF-α, IL-6, and MCP-1) were significantly increased in response to I/R and reached their maximum at 6 hr of reperfusion, which then declined to baseline by 24 hr of reperfusion (Fig. 2). As Fig. 2 shows, hepatic I/R caused significant elevation of TNF-α at 6 hr of reperfusion, which was declined at 24 hr of reperfusion. This data paralleled plasma ALT data, which showed at 6 hr after ischemic insult, followed by a decrease in levels until 72 hr of reperfusion. A similar pattern was observed in plasma IL-6 and MCP-1 levels.
CD44 expression is up-regulated in the liver after I/R
To determine if the CD44-dependent injury was associated with change in protein levels, Western blot analysis was performed on liver lysates from animals that were subjected to liver I/R (Fig. 3A). Following 60 min of warm ischemia, CD44 protein expression was up-regulated as early as 6 hr after reperfusion and then increased in a time dependent manner up to 24 hr. To quantify the amount of protein, a densitometer was used. The values of CD44 in I/R mice were higher by comparison with the values of CD44 in sham mice. The differences were statistically significant (Fig. 3B).
Hepatocellular injury is related to CD44 and neutrophil recruitment in the liver after I/R
Mice were subjected to 60 min of ischemia followed by reperfusion at the time point of 6 hr and 24 hr. The ischemic liver sections were prepared and stained with H&E and then immunohistochemical staining of CD44 and neutrophils using specific anti-CD44 monoclonal antibody and anti-neutrophil antibody. The histopathologic injury of the liver tissue was evaluated based on sinusoidal congestion, cytoplasmic vacuolization, hepatocellular necrosis, and neutrophil infiltration. The liver sections from the sham-operated mice displayed no necrosis, similar to that of the non-operated control mice (Fig. 4A). Additionally, there was no apparent evidence of hepatic injury due to ischemia alone (i.e., at zero hour of reperfusion; image not shown). However, reperfusion of the ischemic liver induced an extensive hepatocellular necrosis, sinusoidal congestion, and neutrophil infiltration after 6 and 24 hr of reperfusion in wild-type mice (Fig. 4D, G). There was sparing of the periportal areas with progressively increased injury approaching the central vein.
CD44 expression is indicated by dark brown color stain. There was progressively increased CD44 expression approaching the central vein (Fig. 4B, E, H). A similar pattern was observed in neutrophil infiltration. The injury was associated with a marked number of neutrophils infiltrated into the midzonal region of ischemic liver after 6 and 24 hr of reperfusion, which was confirmed with in situ immunohistochemical staining of the neutrophils. Neutrophils are also indicated by dark brown color stain (Fig. 4C, F, I).
Pretreatment of neutralizing antibody to CD44 protects against hepatic I/R injury
To determine if endogenous CD44 contributed to organ damage after hepatic I/R, neutralizing antibody to CD44 was administered to mice that were subjected to warm I/R. Mice received a single intraperitoneal injection of anti-CD44 mAb (100 µg per mouse) or control IgG antibody (100 µg per mouse) at 16 hr before surgery as previously described (15). Sixty minutes of warm hepatic ischemia were followed by 6 and 24 hr of reperfusion. Serum ALT levels were analyzed as a measure of hepatocellular injury. As Fig. 5A, B display, serum ALT levels in the mice with pretreatment of control IgG were significantly increased after 6 and 24 hr of reperfusion. This data paralleled in initial I/R mice experiment. However, the serum ALT levels in the mice with pretreatment of anti-CD44 mAb were declined after 6 and 24 hr of reperfusion. The differences compared with the control IgG treated mice were statistically significant (Fig. 5A, B). Treatment with anti-CD44 mAb resulted in significant protection from hepatic I/R injury at each time point.
Liver histology confirmed the serum ALT estimation of hepatocellular damage. Liver sections from mice that were pretreated by control antibody and anti-CD44 mAb before hepatic I/R injury were stained with hematoxylin-eosin. Severe sinusoidal congestion and hepatocellular necrosis was present in liver tissue from mice that were treated with control IgG, whereas minimal damage was noted in samples from anti-CD44 mAb-treated mice (Fig. 5C, D). Moreover, the disruption of CD44 and subsequent inhibition of neutrophil recruitment was confirmed in ischemic liver after 6 and 24 hr of reperfusion by specific immunohistochemical staining. After 6 and 24 hr reperfusion in hepatic I/R mice, the CD44 expression and neutrophil influx were observed in the liver sections from control antibody treated mice. The other hand, immunostaining for CD44 revealed decreased in hepatic sinusoidal area of anti-CD44 mAb-treated mice compared with given control antibody after I/R (Fig. 6A-D). The infiltration of neutrophils into anti-CD44 mAb-treated mice was impaired relative to given control antibody. This pattern of neutrophil influx was similar to that of CD44 immunostaining pattern (Fig. 6E-H).
DISCUSSION
CD44 is constitutively expressed on leukocytes and parenchymal cells including endothelial, epithelial and smooth muscle cells. During inflammation, CD44 expression is up-regulated on hematopoietic and parenchymal cells (17, 18). Several previous studies have indicated that CD44 is involved in a variety of inflammatory responses. The evidence that CD44 plays a crucial role in a variety of inflammatory diseases includes studies in which administration of anti-CD44 antibodies inhibited inflammation in murine models of collagen- and proteoglycan-induced arthritis, experimental autoimmune encephalomyelitis, and IL-2-induced vascular leak syndrome (19, 20). These suppressive effects are thought to be due to reductions in leukocyte recruitment and activation. This has led to the suggestion that CD44 should be considered as a target for novel interventions in inflammatory diseases. In our study, we showed that CD44 exerted a crucial role in migration of neutrophils into the postischemic tissue from I/R. The CD44 blockade was shown to decrease neutrophil influx, reduce hepatic I/R injury, and preserve hepatic function.
First, we showed that CD44 was rapidly up-regulated in postischemic tissue. Although early after I/R injury, its expression was found on infiltrating leucocytes and sinusoidal endothelial cells, the CD44 expression actually was increased until 24 hr after I/R injury. These findings are in agreement with observations in testicular I/R injury. Moon et al. (21) showed that the expression of CD44 significantly increased and that abundant number of inflammatory cells infiltrated into the interstitial space surrounding damaged semiferous tubule during the delayed phage (24-96 hr) after testicular I/R injury. Therefore, we can postulate that the CD44 leads to increase neutrophil-dominated liver injury during the later phase after hepatic ischemia, as described earlier by Jaeschke (22).
Second, we demonstrated that disruption of CD44 attenuated hepatic injury after I/R and preserved hepatic function. Generally, the I/R injury is characterized by recruitment and infiltration of neutrophils into the postischemic tissue and accentuated by the release of cytotoxic proteases and oxygen-derived free radicals (23). In accordance, depletion of neutrophil recruitment into the postischemic tissue by blockade of cellular adhesion molecules decreases hepatic I/R injury (24). In our study, we found that infiltration of neutrophil into the postischemic tissue decreased significantly in CD44 mAb pre-treatment group. Our data agree with other animal model, including renal I/R injury (25), in which administration of an antibody that interact with the hyaluronan (HA)-binding site of CD44 induced shedding of CD44 and decreased neutrophil migration into the postischemic kidney. Moreover, injection of the antibody did not alter peripheral blood leukocyte numbers or neutrophil, lymphocyte, or monocyte counts. McDonald et al. (8) recently demonstrated that interaction of CD44 and HA is the dominant mechanism for neutrophil sequestration in inflamed liver sinusoids of mice in response of LPS (or bacterial endotoxin)-induced hepatic injury. Since neutrophil recruitment occurs quite rapidly in for example endotoxemia, it stands to reason that constitutively expressed or presynthesized molecules would be important in neutrophil recruitment into tissues. They suggested that CD44-mediated adhesion of neutrophils was independent of selectins. Then, RHAMM-1, another HA receptor, played no role in neutrophil adhesion in sinusoids. Moreover, they showed that hyaluronidase, an HA depleting enzyme, CD44-/- mice and anti-CD44 antibody all reduced neutrophil adhesion in sinusoids and reduced tissue injury. Therefore, we can suggest that CD44 is considered as a target for novel interventions in hepatic I/R injruy.
The significance of this study is the first description that the CD44, selectin-independent specific cellular adhesion molecule on neutrophil or endothelium is important to pathophysiological process in the liver from I/R injury. We hypothesized that CD44 exerts a crucial role of the migration of neutrophils into the postischemic tissue from I/R. However, there are some points that we were not entirely satisfied to elucidate the role of CD44 in hepatic I/R injury. The most clinically relevant findings reported in this study were the observation that improved hepatic function and decreased neutrophil infiltration into the postischemic tissue was ameliorated by administration of an anti-CD44 mAb.
To determine whether our results could be valid for therapeutic application, further studies are necessary to examine accurate mechanism of CD44-HA interaction leading to neutrophil recruitment from hepatic I/R injury. The involvement of CD44 in migration of neutrophils in extracellular matrix may vary according to the distribution of HA contents (26). Therefore, the demonstration of HA at the cell surface of murine neutrophils is an important for elucidating mechanism of CD44-HA combination from hepatic I/R injury. Secondly, future studies are necessary to examine the specific role of CD44 in recruitment of neutrophil from hepatic I/R. Khan et al. (27) showed that rolling of leukocytes in chronic inflammation by intrascrotally injected with the neutrophil-activating chemokine was not impaired in absence of CD44. This suggests that CD44 plays a specific role in the adhesion of neutrophil. Finally, our study is in part consistent with wild type data, in that the up-regulation of the CD44 correlated with hepatic injury and neutrophil infiltration during later-phage of hepatic I/R (i.e. 24 hr of reperfusion). However, previous studies have suggested that the neutrophil recruitment may haven mediated by other more potent chemoattractants and mediators (28, 29). Using wild-type and IL-1 receptor I-knockout (IL-1RI[-/-]) mice, Kato et al. (30) also showed that IL-1RI(-/-) mice had significantly less neutrophil accumulation in hepatic I/R. Thus, using CD44-/- or chimeric mice, future studies are necessary to confirm the potential role of CD44 in neutrophil recruitment from hepatic I/R.
In conclusion, up-regulation of CD44 on sinusoidal endothelial cells after I/R injury results in the recruitment of neutrophils into the postischemic tissue. The disruption of CD44 via neutralizing antibody to CD44 resulted in down-regulation of CD44, decreased influx of neutrophils and attenuation of hepatic I/R injury. Therefore, targeting CD44-HA interactions may be an innovative and efficient therapy to prevent or reduce I/R related hepatic injury.





 XML Download
XML Download