

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pancreatoblastoma is a rare primary pancreatic neoplasm of children. It was first described in a 15-month-old boy as "infantile adenocarcinoma of the pancreas" by Becker (1) in 1957. Pancreatoblastoma usually affects children between 1-8 yr, although rare cases in adults have been reported (2). There is a male predominance and more than half of the reported cases are in Asians (3, 4). The tumor may arise from any portion of the pancreas; however, a tumor that originates from ectopic pancreatic tissue is extremely rare. To the best of our knowledge, a single case of a 4.3-yr-old boy with a tumor originating from ectopic pancreatic tissue (mesentery) has been reported by Yang (5), however, cases with synchronous ectopic pancreatoblastoma have only been reported recently. Although the radiologic features of pancreatoblastoma are well described in the literature, few reports describe ectopic pancreatoblastoma, particularly about synchronous ectopic pancreatoblastoma. We present a case of a 3-yr-old boy with ectopic pancreatoblastoma originating from the omentum, who has remained disease free more than 1 yr after complete tumor resection.
CASE DESCRIPTION
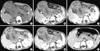
A 3-yr-old boy presented with abdominal pain was referred to our institution for the evaluation of a progressive bulge in his right abdomen that his mother had first noticed seven days previous on February 12, 2009. A biochemical evaluation revealed no significant abnormalities. Serum levels of tumoral markers (AFP, CA19-9, CA125 and CEA) were within reference limits. A physical exam revealed a mildly tender mass in the right hypochondrium extending to the epigastrium; however, two lesions were found after a dynamic computed tomography (Fig. 1). The primary lesion (8.5 × 4.3 cm) was located in the head of the pancreas and partly herniated into the abdominal wall. While the secondary lesion (1.8 × 2.1 cm) was in the left abdomen near the spleen and the tail of pancreas, and well demarcated from the surrounding structures, and no calcifications were evident within them (Fig. 1A). They all showed well-defined margins and inhomogeneous attenuation due to the presence of cystic and solid areas, in the venous phase (Fig. 1C) and the delayed phase (Fig. 1D, E), a continuous enhancement was observed with a parenchyma density that was higher than that of the arterial phase. In addition, a large and distorted vascular shadow (Fig. 1B) inside of the tumors in the arterial phase in the primary lesion. There was no radiological evidence of nodal or distant metastases; however, there was a local infiltration of the adjacent abdominal muscle that required a laparotomy.
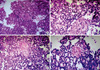
Surgery revealed the primary lesion was firmly adhered to the adjacent omental and abdominal muscle; in addition, a local infiltration of the liver and stomach were also noted. The head of the pancreas was compressed and could be separated from the tumor due to well-defined margins. The bile duct, portal vein, and the inferior vena cava were also compressed but patent. The gross specimen showed a resected mass measuring 8 × 4 × 4 cm that was well circumscribed and solid with areas of necrosis and hemorrhaging. No calcifications were evident within the mass. The secondary lesion was also adhered to the adjacent omentum and spleen; however, it was well demarcated from the surrounding structures with no evidence of originating from the pancreas. A histopathological analysis revealed a resected mass measuring 3 × 4 × 3 cm, which was well encapsulated by omentum with no calcifications within the mass. The surgical resection was difficult due to the extensive nature of the tumor, but the two lesions were completely resected without the pancreas being damaged. In the final pathology report following surgery, the diagnosis of pancreatoblastoma was confirmed with histopathological results, and the secondary lesion histology and immunohistochemistry was consistent with the findings of the primary lesion (Fig. 2).
After surgery, the patient was treated with adjuvant chemotherapy (vincristine, cyclophosphamide, and epirubicin) regularly. At the time of this report, 18 months after complete tumor resection, the patient is currently in good condition and without radiological evidence of recurrent tumor or metastases (Fig. 1F) with normal levels of CA 125, CA 19-9 and AFP to date.
DISCUSSION
Ectopic pancreas, which is also known as heterotopic or aberrant pancreas, describes pancreatic tissue in an aberrant location without vascular or ductal connection to the orthotopic pancreas (6). Ectopic pancreas is a relatively frequent congenital anomaly and the reported incidence ranges from 2% to 15% (7); however, the true incidence is unknown as most patients are asymptomatic and the condition is usually an incidental finding during autopsy or laparotomy. The most frequent sites are the gastric antrum (30%), duodenum (30%), and jejunum (20%) (7). On occasion, an ectopic pancreas has been found in the ileum, colon, spleen, liver, umbilicus, biliary tract, omentum, mesentery, mediastinum and Meckel's diverticulum (8, 9). Ectopic pancreas occurs in all ages and the male-to-female ratio is approximately 2:1. The histogenesis of this lesion is still unclear. Ectopic pancreas undergoes changes similar to the native pancreatic gland and symptoms often depend upon location. Changes to the ectopic tissue can include acute or chronic pancreatitis, fibrosis, and even the development of pancreatic adenocarcinoma (10, 11).
Pancreatoblastoma accounts for 0.5% of exocrine tumors of the pancreas and the highest incidence is found in the first decade of life with a predilection for males and Asians (3, 4). Pancreatoblastoma may arise in any portion of the pancreas. Pancreatoblastoma originating from the ectopic pancreas is extremely rare, a single case of a 4.3-yr-old boy with a tumor originating from ectopic pancreatic tissue (mesentery) has been reported so far (5). In our study, we found a 3-yr-old boy with two synchronous pancreatoblastoma in the abdomen. A laparotomy revealed that the head of pancreas was compressed but normal. Serial postoperative radiological examinations also revealed no mass in the pancreas. In the final pathology report following surgery, the diagnosis of pancreatoblastoma was confirmed, and the secondary lesion histology and immunohistochemistry was consistent with the findings of the primary lesion. Guillou et al. (12) described 3 conditions necessary to prove that a malignancy arose from ectopic pancreas: 1) the tumor must be within or near the ectopic pancreatic tissue, 2) a direct transition between pancreatic structures and carcinoma must be observed, and 3) the non-neoplastic pancreatic tissue must at a minimum comprise fully developed acini and ductal structures. Our case satisfied all 3 conditions. Therefore, we believe that the two synchronous tumors may originate from ectopic pancreatic tissue in the omentum there has been unreported in previous studies. It supported the view that the pancreatoblastoma was embryonic in origin and hamartomatous in the early stages of the embryo.
The pathologic features of pancreatoblastoma are well described in the literature, but few series describe the radiologic features of pancreatoblastoma. The radiological features are usually those of well-defined, lobulated, heterogeneous masses, with necrosis and/or calcification, usually arising from the body and/or tail of the pancreas or involving the entire organ, rather than being universally located in the pancreatic head (13). The mass is often so large at presentation as to make determination of the organ of origin quite difficult. In the series of Montemarano et al. (14), in only half of the cases did the imaging appearance suggest the pancreas as the organ of origin. These large tumors typically compress the surrounding structures without appearing to invade them, although local invasion may be evident at surgical resection (14, 15). In our case, the two tumors were all heterogeneous and well-defined; in addition, calcification were not seen within them. In the arterial phase, a moderate enhancement was observed in parenchyma, while in the venous phase and delayed phase, continuous enhancement was observed with a parenchyma density higher than that of the arterial phase. In addition, a large and distorted vascular shadow inside the tumors in the arterial phase was in the primary lesion. Despite the fact that CT findings suggest an encapsulated tumor, the local infiltration of the liver and stomach were also noted at surgical resection.
Although there are certain characteristic imaging findings, the diagnosis of pancreatoblastoma relies mainly on pathology. The presenting features of pancreatoblastoma are commonly nonspecific and referable to the presence of an upper abdominal mass. Since pancreatoblastomas are of a soft and gelatinous consistency, they rarely cause biliary or duodenal obstruction, but they may encase adjacent vessels. More than 15% of patients with pancreatoblastoma present with metastases at the time of diagnosis and additional patients develop metastases later in the course of the disease (4). The liver is the most common site of metastases; however, regional lymph nodes, peritoneal, bone, pulmonary, and mediastinal metastases have also been described (16, 17). Vascular invasion is uncommon and ascites may be an indicator of tumor spread (14). According to the biological growth characteristics of pancreatoblastoma and to the literature review, complete surgical resection remains the treatment most commonly associated with a good prognosis and no late complications. Unresectable or recurrent disease was shown to respond to chemotherapy. The production of alpha-fetoprotein (AFP) by pancreatoblastoma has been noted repeatedly in the literature, both in the form of serum elevation and immunohistochemical detection. AFP may be useful for diagnosis and used as a tumor marker in follow-up after surgery and during chemotherapy (18). It also should be noted that the value of serum AFP is limited in neonates, because a higher level of serum AFP may be normal. Our patient had two localized tumors that were completely resected and the child is currently doing well and 1.5 yr disease-free at follow-up. Preoperative and postoperative serum levels of tumoral markers (AFP, CA19-9, CA125 and CEA) were within reference limits.
In conclusion, pancreatoblastoma is a rare primary tumor of the pancreas and ectopic pancreatoblastoma is even rarer. When a large well-defined, lobulated, heterogeneous mass, with necrosis and/or calcification is identified in the pancreas of children, the diagnosis of pancreatoblastoma should be considered. Furthermore, an ectopic pancreatoblastoma should be included in the differential diagnosis when identified within or near the ectopic pancreatic tissue. As the present case shows, this tumor can also occur synchronously outside the orthotopic pancreas: synchronous ectopic pancreatoblastoma. We report on this rare disease to emphasize its clinical features, radiological findings, laboratory findings, diagnosis, and management. To our knowledge, synchronous ectopic pancreatoblastoma was reported for the first time.
 XML Download
XML Download