

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatocellular carcinoma (HCC) is the fifth most common cancer worldwide and a rise in the incidence and mortality from HCC has recently been observed in most industrialized countries (1). At the time of presentation, less than 40% of HCC patients are eligible for potential curative treatment (2) and, unfortunately, conventional systemic chemotherapeutic agents such as doxorubicin, 5-fluorouracil and cisplatin do not have significant efficacy in HCC, based on data from randomized trials (3). With the progress in the knowledge of tumor biology, new targeting therapies blocking specific signal pathway have been developed recently in many solid and hematologic malignancies and there is a great need to find out novel targets and to develop specific blocking agents to these targets in HCC as well. Hepatitis B virus (HBV) infection is a major risk factor for the development of HCC and accounts for the high incidence of HCC in Asian and African countries including Korea (4, 5). Hepatitis B virus X (HBx) protein, which is encoded in HBV open reading frame X gene, has been suggested to play an important role in the development of HCC in the setting of chronic HBV infection (6). Expression of HBx protein in HCC cells results in the increased β-catenin activity (7, 8), activation of Ras/Raf/MEK/ERK pathway (9) and PI3K/Akt pathway (10, 11) which are potential targets for developing new therapeutic agents.
Ras/Raf/MEK/ERK pathway is one of the most critical signaling cascades for liver tumorigenesis (12). This pathway is central in cell growth and survival, transducing extracellular signals from ligand-bound tyrosine kinase receptors such as epidermal growth factor receptor to the cell nucleus through a series of specific phosphorylation events that start with the activation of Ras. Although EGFR is not commonly overexpressed in HCC cells, its continuous activation by ligands such as transforming growth factor-α (TGF-α), heparin-binding epidermal growth factor or amphiregulin, which are upregulated from the early stages of hepatocarcinogenesis, can be of significance for HCC development. The inhibitory activity of epidermal growth factor receptor tyrosine kinase (EGFR-TK) inhibitor such as gefitinib and erlotinib on human HCC cell growth in culture has been recently demonstrated (13, 14). These compounds induced cell cycle arrest and apoptosis, and erlotinib enhanced chemosensitivity towards cytostatics (14). In addition, in vivo studies have shown that gefitinib displays antitumoral effects in a rat model of chemically induced liver cirrhosis and HCC (15). However, this antiproliferative action of gefitinib on HCC cells was suggested to be effective only when TGF-α/EGFR autocrine loop and the following downstream pathway of EGFR are intact in HCC cells (16). HBx protein expressed in HBV infected hepatocyte was reported to activate MAPK pathways irrespective of EGFR activation through Src kinase activation in several in vitro and in vivo studies (9, 17). Therefore, for the treatment of HCC's in Asian countries, where HCC is highly prevalent and HBV infection is the main cause of carcinogenesis in hepatocytes, the antiproliferative efficacy of EGFR-TK inhibitor on HCC cells could be limited. On the contrary, downstream pathway inhibitors, such as MEK inhibitors could be more appropriate candidates for the growth inhibition of HCC cells expressing HBx protein and HCC patients infected with HBV in this aspect.
To confirm this hypothesis, our experiments were aimed to 1) establish HCC cell lines which are stably transfected with HBx gene, 2) confirm the effect of HBx expression on the several signal pathways such as Ras/Raf/MEK/ERK, Wnt/β-catenin, and PI3K/Akt pathway and, 3) eventually, compare the antiproliferative efficacy of EGFR-TK inhibitor and MEK inhibitor in these cells.
MATERIALS AND METHODS
HCC cell lines and HBx gene transfection
Human HCC cell lines (HepG2, Huh-7) were purchased from Japanese Collection of Research Bioresources (JCRB). Both cell lines were cultured in Dulbecco modified Eagle medium (DMED) supplemented with 10% fetal bovine serum, 100 units/mL penicillin and 100 mg/L streptomycin.
To establish HCC cell lines expressing HBx protein, plasmid with HBx gene was transfected into HepG2 and Huh-7 cells using Lipofectamine 2000 reagent in Opti-MEM (Invitrogen, Carlsbad, CA, USA) according to the manufacture's protocol. HBx (subtype ayw) expressing plasmid vector (pEG-HBx) and pEGFP-N1 (negative control) were kindly provided by Dr. Kyun-Hwan Kim (Konkuk University, Seoul, Korea). For stable cell lines, the cells were maintained in selective growth medium supplemented with 600 µg/mL G-418 (Sigma, St. Louis, MO, USA) after transfection. The expression of HBx gene was confirmed by reverse transcription-polymerase chain reaction (RT-PCR) and immunofluorescence assay.
Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was extracted from cultured cells using RNA Purification System Mini Kit (Invitrogen) and cDNA was amplified using the Superscript II reverse transcriptase system (Invitrogen) and the pfu PCR pre-Mix kit (Bioneer, Daejeon, Korea). The HBx gene was amplified using forward 5'-CAT GGC TGC TAG GCT GTG CTG-3' and reverse 5'-GAG ATG ATT AGG CAG AGG TGA AAA AG-3' primers (18). The size of PCR product was 473 bp for HBx gene. PCR products were loaded on a 1.5% agarose gel with ethidium bromide and image was obtained by photo image analyzer (Bio Rad, Hercules, CA, USA).
Immunofluorescence staining
Cells were plated in two-chamber glass slides at a density of a 2.0 × 104 cells per well. After 24 hr in culture medium, the cells were washed three times with phosphate-buffered saline (PBS) and then fixed with 3.5% paraformaldehyde solution. The cells were permeabilized with 0.1% Triton X-100 and nonspecific binding was blocked with 5% bovine serum albumin in PBS. Subsequently, cells were incubated with the primary monoclonal HBx antibody (Chemicon, Temecula, CA, USA) overnight and exposed to anti-mouse IgG conjugated Alexa Fluor 546 (Invitrogen) in a dark chamber for 1 hr. For the negative controls, other slides were incubated with the same buffer without the primary monoclonal HBx antibody. All the slides were mounted with mounting medium containing DAPI (Vector Laboratories, Burlingame, CA, USA) and viewed under a fluorescence microscope (Leica microsystems, Nussloch, GmbH, Germany).
EGFR-TK and MEK inhibitor treatment
Gefitinib (EGFR-TK inhibitor) and selumetinib (AZD6244; MEK inhibitor) were kindly provided by AstraZeneca Pharmaceuticals. Stock solutions were prepared at 20 mM in dimethyl sulfoxide (Sigma) and stored in aliquots at -20℃. In immunoblotting assay, these inhibitors were treated for 20 hr in each cell line before protein collection. In cell proliferation assay, these inhibitors were treated from day 0.
Immunoblotting
Cells were lysed in radioimmune precipitation (RIPA) buffer (Upstate, NY, USA) supplemented with protease inhibitor. The cell lysates were electrophoresed on 10% polyacrylamide gel, transferred onto polyvinylidene difluoride membrane (Bio-Rad Laboratories) and blotted with appropriate primary and secondary antibodies. The signal was detected using ECL reagent kit (Biosciences, Buckinghamshire, UK) and exposed to an X-ray film. The primary antibody include: anti-pERK (Thr202/Tyr204), anti-pAKT (Ser473), anti-β-catenin (all from Cell signaling Technology Inc., Beverly, MA, USA), anti-β-actin (Sigma). Goat anti-rabbit IgG antibody conjugated with horseradish peroxidase was used as the secondary antibody (Santa Cruz, CA, USA).
Reporter gene assay
Cells were cotransfected over 24 hr using 20 ng TK Renilla-CMV and 0.2 µg TCF reporter plasmid (Upstate Biotechnology Inc., Lake Placid, NY, USA). Firefly and Renilla luciferase activities were quantified using a dual luciferase reporter assay system (Promega, Madison, WI, USA). Data were expressed as ratios of firefly to Renilla luciferase activity. The experiment was repeated at least three times for each cell line.
Cell proliferation assay
Cell proliferation was measured using the CellTiter 96 Aqueous One Solution cell proliferation assay (Promega), on the basis of the cellular conversion of the colorimetric reagent MTS (3,4-[5-dimethylthiazol-2-yl]-5-[3-carboxymethoxyphenyl]-2-[4-sulfophenyl]-2H-tetrazolium salt) into soluble formazan by dehydrogenase enzymes found only in metabolically active, proliferating cells. Following each treatment, 20 µL of dye solution was added into each well in 96-well plate and incubated for 3 hr. Subsequently, absorbance at 490 nm was measured with an ELISA reader (Magellan, TECAN, Austria).
Primary culture of HCC cells
We performed primary culture of HCC cells from surgically excised HCC tissue. The study protocol was in accordance with the ethical guidelines of the 1995 Declaration of Helsinki and was approved by the Investigation and Ethics Committee for Human Research (protocol No. 2007-0332) at Asan Medical Center. An informed consent was obtained from all the patients before surgical resection. After excision of HCC tissue from patients, 1 cm3 of HCC tissue was transferred immediately to Hank's balanced salt solution (HBSS) free of calcium and magnesium (19). The liver was gently dispersed and chopped with surgical knife and placed in HBSS containing 0.03% pronase, 0.05% type IV collagenase, and 0.01% deoxyribonuclease (DNase, from bovine pancreas) (all from Sigma) for 20 min at 37℃. The resulting cell suspension was filtered through a 100 µm-nylon filter (Falcon, Becton Dickinson, Franklin Laces, NJ, USA) and centrifuged at 50 g for 2 min at 4℃ to pelletize the hepatocytes. The pellet was resuspended in DMEM supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 mg/L streptomycin and seeded in uncoated plastic culture dishes (100 mm Petridish, Primaria, Falcon) at ca. 1 × 106 cells/mL. The medium was replenished 12 and 24 hr after plating and every 48 hr thereafter. After over 100 primary cultures, we got the one cell line, which grows over 40 passages of culture, and performed cell proliferation assay with gefitinib or selumetinib in this cell.
RESULTS
Establishment of HBx protein expressing HCC cell lines
We transfected pEG plasmid containing HBx gene (pEG-HBx) into HepG2 and Huh-7 cells and established stably transfected cell lines. HBx gene expression was confirmed by RT-PCR in these cells (Fig. 1A). This expression of HBx mRNA was stably sustained over 20 passages of culture in these cells. We also confirmed HBx protein expression in transfected cell lines by immunofluorescence staining. DAPI staining indicates the site of nucleus and GFP fluorescence showed the transfected HCC cells. Compared with vector plasmid transfection, pEG-HBx transfected HCC cells showed strong HBx protein expression in immunoflurescence staining (Fig. 1B, C).
pERK and pAkt expression in HepG2 and Huh-7 cells and β-catenin activity were enhanced in Huh-7 cell by HBx transfection
In Huh-7 cells, the expression of pERK was constitutively elevated and further activated by HBx transfection. HepG2 cells also showed the enhancement of pERK expression by HBx transfection but the expression level was weaker than Huh-7 cells. Treatment of TGF-α activated pERK expression in both cells as previously reported (Fig. 2A). pAkt expression was also activated by HBx transfection and TGF-α treatment in both cell lines (Fig. 2B, C). HBx transfection enhanced β-catenin activity significantly in Huh-7 cells, which was confirmed by reporter gene assay (P < 0.05) (Fig. 2D), but the expression of β-catenin was not affected by either HBx transfection or TGF-α treatment (Fig. 2B, C).
Effect of gefitinib on the expression of pERK, pAkt, β-catenin in HBx transfected HCC cell lines
Gefitinib inhibited effectively HBx transfection and/or TGF-α treatment induced pERK and pAkt expression in HepG2 cells. Gefitinib also inhibited expression of pERK and pAkt in Huh-7 cells but this inhibition was not complete. HBx transfected Huh-7 cells showed stronger expression of pERK than control cells even after the treatment of gefitinib (Fig. 2B). β-catenin expression was not affected by gefitinib treatment in both cell lines but β-catenin activity was significantly inhibited by gefitinib in Huh-7 cells regardless of HBx transfection (Fig. 2D).
Effect of selumetinib on the expression of pERK, pAkt, β-catenin in HBx transfected HCC cell lines
Selumetinib strongly inhibited the enhancement of pERK expression by HBx transfection and/or TGF-α treatment in both HepG2 and Huh-7 cells. pAkt expression was rather elevated after treatment of selumetinib in both cell lines (Fig. 2C). The level of β-catenin expression was not changed by selumetinib in both cell lines but β-catenin activity was significantly inhibited by selumetinib regardless of HBx transfection and the suppression was stronger than gefitinib in Huh-7 cells (Fig. 2D).
Cell proliferation inhibition by gefitinib or selumetinib in pEGFP-N1 or pEG-HBx transfected HepG2 and Huh-7 cell lines
Gefitinib 10 µM inhibited the growth of HepG2 and Huh-7 cell lines effectively regardless of HBx transfection. HBx transfection did not affect antiproliferative effect of 10 µM gefitinib in both cell lines. However, gefitinib 1 µM did not show significant growth inhibition in both cell lines irrespective of HBx transfection (Fig. 3). Selumetinib inhibited the growth of HepG2 and Huh-7 cells profoundly in a dose dependent manner. The growth inhibition by selumetinib was not influenced by HBx transfection in either cell line (Fig. 4). However in view of the fact that concentrations in excess of 1 µM are required to slow down cell growth, these cells can be considered to be resistant to MEK inhibition.
Cell proliferation inhibition by gefitinib or selumetinib in primary cultured HCC cell line
We performed primary culture with 100 HCC tissues resected from HCC patients and one cell line, which came from HCC tissue resected from a female patient with HBV infection, has become established after serial passage. These cells grew stably over 50 passages without gross morphological changes. We performed cell viability assay with gefitinib or selumetinib in these cells. Gefitinib 10 µM inhibited the growth of primary cultured HCC cell line effectively but 1 µM did not show such inhibitory effect. Selumetinib inhibited the growth of these cells profoundly even in a dose of 1 µM (Fig. 5) which therefore seem somewhat more sensitive to selumetinib than either of the established cell lines.
DISCUSSION
Because conventional cytotoxic chemotherapy has not been shown to prolong survival in unrespectable HCC patients (3), there is a significant unmet need for the new therapies to prolong survival of HCC patients who are not eligible for potential curative treatment. With the progress in the knowledge of hepatocarcinogenesis, several signal pathways, such as Ras/Raf/MEK/ERK, PI3K/Akt/mTOR, Wnt/β-catenin and vascular endothelial growth factor (VEGF) activity, were explored extensively in HCC cells and many molecular targeting agents which inhibit specific step of each pathway were found to have potential role in the treatment HCC in in vitro or in vivo experiments and early phase clinical trials (20). Recently, significant progress has been made in the study of molecular targeting agent for HCC. Sorafenib, which inhibits both Raf kinase and VEGF receptor, has been shown to prolong survival of HCC patients in phase-III clinical trial (21). However the partial response rate of sorafenib was only 2%; and novel molecular targeting agents or new combination regimens are still necessary in the treatment of HCC.
EGFR-TK inhibitor (gefitinib) and MEK inhibitor (selumetinib) are other potentially promising agents in HCC treatment because EGFR/Ras/Raf/MEK/ERK pathway is one of the most critical signaling cascades for hepatocarcinogenesis. In in vitro experiments, gefitinib showed anti-proloferative effect in several HCC cell lines (13-15) but the growth inhibition was effective only when TGF-α/EGFR autocrine loop and the following downstream pathway of EGFR are intact in HCC cells (16). Selumetinib was reported recently to have antiproliferative effect in HCC cell lines in one report (22).
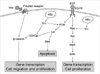
HBx protein, which is expressed in HCC cells of chronic hepatitis B patients, can activate the three signal pathways (7-11). Therefore, we postulated that the antiproliferative effect of gefitinib and selumetinib could be different according to the expression of HBx protein in HCC cells because HBx expressing HCC cells could bypass EGFR pathway to activate MEK through HBx/Src kinase activation (9, 17) (Fig. 6). Therefore, in our experiments we aimed to find out the effect of HBx transfection on the antiproliferative effects of gefitinib and selumetinib in HCC cell lines.
We established stably HBx transfected HepG2 and Huh-7 cell line. In other several reports (8-11), HBx protein expression was confirmed with Western blotting band but in our experiment the band of this small protein (17 kDa) could not be obtained. Instead, we confirmed HBx expression with immunofluorescence staining. pERK and pAkt expression and β-catenin activity were activated by HBx transfection in Huh-7 and to a lesser extent in HepG2 cells as in previous reports (7-11). Gefitinib inhibited ERK phosphorylation in both cells regardless of HBx transfection but, in Huh-7 cells, HBx transfected cells showed stronger pERK expression than control cells even after gefitinib treatment. In contrast, selumetinib inhibited ERK phosphorylation profoundly in both HepG2 and Huh-7 cells regardless of HBx transfection, which was in accordance with our hypothesis. Regarding pAkt, gefitinib inhibited pAkt expression effectively in both cells regardless of HBx transfection, but treatment of selumetinib lead to some increase in the expression of pAkt in both cell lines. These findings suggest that the cross-talk between EGFR/Ras/Raf/MEK/ERK and PI3K/Akt/mTOR pathway could be in the level of EGFR/Ras/Raf and the antiproliferative effect of selumetinib on HCC cells could be compensated by PI3K/Akt/mTOR pathway activation in these cells. And also, gefitinib could have more potent antiproliferative effect by inhibiting both EGFR/Ras/Raf/MEK/ERK and PI3K/Akt/mTOR pathways in HCC cells. Regarding β-catenin activity, selumetinib showed stronger inhibition than gefitinib in both cells regardless of HBx transfection.
According to our hypothesis, we expected that the antiproliferative effect of gefitinib was limited in HCC cell lines transfected with HBx gene compared with control cells and the antiproliferative effect of selumetinib was not affected by HBx transfection in HCC cells. However, antiproliferative effect of both agents was not affected by HBx transfection, indeed both cell lines were relatively resistant to selumetinib and gefitinib. So, the effect of HBx transfection on each pathway was confirmed respectively in our experiments but the functional assay failed to prove our hypothesis. Considering the complex network of signal pathways (20), there is a possibility that the activation of pERK, pAkt and β-catenin by HBx transfection was not strong enough to attenuate the antiproliferative effect of gefitinib and moreover, gefitinib could inhibit PI3K/Akt/mTOR pathway as well as EGFR/Ras/Raf/MEK/ERK in our experiment. Further experiments with more trial and error will be needed to find out the most effective targeting agent or combination regimen in the treatment of HCC.
In conclusion, HBx expression enhanced pERK, pAkt and β-catenin activity in HepG2 and Huh-7 cell lines. EGFR-TK inhibitor could inhibit pERK expression in both HCC cell lines but this inhibitory effect was attenuated by HBx expression in both cells. However, antiproliferative effect of EGFR-TK inhibitor was not affected by HBx expression in both cell lines, and it might be explained by the other hypothesis that the activation of signal pathway by HBx transfection could not be strong enough to attenuate the antiproliferative effect of gefitinib, or gefitinib could show antiproliferative effect by inhibiting both EGFR/Ras/Raf/MEK/ERK and PI3K/Akt/mTOR pathway in HCC cell lines. Targeted inhibition of MEK activity by selumetinib may represent a valuable alternative approach for the treatment of HCC. Future experiments will be needed to understand the role of HBx protein expression in the treatment of HCC while blocking relevant pathway by molecular targeting agent, and to find out the most effective targeting agent or combination regimen considering the complex network of signal pathways in hepatocarcinogenesis.





 XML Download
XML Download