

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The rare association between β-thalassemia and mental retardation was first recognized in 1981 by Weatherall and colleagues (1). Then, in 1995, in a case of β-thalassemia/mental retardation (ATR-X) syndrome, mutations in the ATRX gene locus Xq13.3 were first reported (2). Since then, studies have identified 127 mutations in this gene in patients with ATR-X syndrome (3-7). While the cellular function of ATRX has not yet been fully elucidated, the characteristics of the syndrome suggest that ATRX probably functions as part of a chromatin remodeling complex to regulate the transcription of a discrete set of target genes, of which β-globin genes involved in brain development are the best characterized example (8-10).
Although there have been many studies and case reports of this syndrome and the ATRX gene all over the world, no case has been reported in Korea until now. We encountered the first Korean case of ATR-X syndrome and reported it.
CASE DESCRIPTION
A 32-month-old boy was admitted to our hospital with two days of irritability and fever. He was born with a birth weight of 2.9 kg by normal spontaneous vaginal delivery at 40 weeks gestation to a healthy 28-yr-old G3P2 mother. His delivery was non-eventful, but there was flexion deformity of both his middle fingers, so he was transferred to our hospital and underwent many diagnostic tests and physical therapies in January, 2002. He showed mild general hypotonia in that his cry was weak, and he sucked poorly during early infancy. He had epileptic seizures 6 times since the age of 15 months despite taking anti-epileptic medications. A diagnosis of mental retardation complicated by epilepsy was made, and his development was noted to be markedly delayed. He raised his head at age 7 months, sat up at 26 months, and had not yet acquired the ability to stand. He had a flat and mid-hypoplastic face with prognathism, narrow and upward slanting palpebral fissures with hypertelorism, low-set ears, a small crashed nose, widely spaced incisors, carp-like mouth, and round back (Fig. 1). He demonstrated repeated stereotyped behavior like hitting his chin with his palm; this behavior was often associated with emotional outbursts.
Soon after admission, a generalized tonic-clonic type seizure was started and lasted approximately one hour; at that time his vital signs were as follows: blood pressure 100/60 mmHg, pulse rate 138 bpm, respiration rate 32 bpm, and body temperature 38.5℃. Mild throat injection was seen, but other systemic examinations were normal. On neurologic exam, mental status was alert, all cranial nerve exams were normal, and muscle tone in both extremities were decreased to grade IV/IV bilaterally but all deep tendon reflexes were physiologic. There was no spasticity or pathologic reflex. Sensory functions were normal and meningeal irritation signs were absent.
Initial laboratory investigations revealed: hemoglobin 12.7g/dL, platelet count 374,000/µL and white blood cell count 11,780/µL. The C-reactive protein was slightly elevated at 19.0 mg/dL on the day of admission and returned to normal 3 days later. Urinalysis was normal. All microbiologic studies were negative. A sleep electroencephalogram revealed intermittent high-amplitude slow wave discharges from the temporo-occipital area, representing mild cerebral dysfunction. On radiologic evaluation, spine radiography revealed mild scoliosis. An echocardiogram was normal.
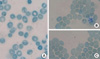
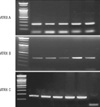
The characteristic facial features and mental retardation of the patient led us to consider ATR-X syndrome; thus, peripheral RBCs were screened for hemoglobin H (HbH) inclusion bodies. HbH inclusions were detected under the microscope in 1.1% of brilliant cresyl blue stained RBCs, consistent with the diagnosis of ATR-X syndrome (Fig. 2). The patient's father, mother and two sisters were all given the same test; only his mother and eldest sister had the same findings. We performed gene analysis to confirm the diagnosis of ATR-X syndrome. Genomic DNA from the peripheral blood of our patient and his 4 family members was extracted with the Wizard® Genomic DNA Purification Kit (Promega, Madison, WI, USA) and used as a template for amplification of the ATRX gene. Polymerase chain reaction (PCR) was separated into three parts and carried out with sequence-specific oligonucleotide primers containing 20 known major mutation sites (Table 1, Figs. 3, 4). PCR amplification using each primer was conducted with premixture kit (PreMix™-Top, Bioneer Inc., Seoul, Korea). Cycling conditions were one cycle at 95℃ for 5 min, followed by 35 cycles of 95℃ for 30 sec, at an annealing temperature adjusted to each primer's (Tm) for 30 sec, 72℃ for 30 sec, followed by one cycle at 72℃ for 7 min. For direct sequencing, the PCR products were purified using a PCR clean-up system offered by Promega. DNA sequencing was performed using the purified PCR products as template, the same primers as those used for template generation, the BigDye terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA, USA), and automatic sequencer ABI Prism 3730 XL DNA Analyzer (Applied Biosystems, Foster City, CA, USA). Sequence data were assembled and compared to that of a previously reported ATRX sequence from accession number Z84487 using the DNAstar software (DNASTAR Inc., Madison, WI, USA).
Mutation analysis for our patient showed a point mutation on the 9th exon in the ATRX gene of thymine to cytosine so that Trp(C), the 220th amino acid, was replaced by Ser(R) (Fig. 5). This missense mutation was reported by Gibbons in 1997 (8). We investigated the same mutation in his 4 family members, and his mother and two sisters were found to be carriers.
DISCUSSION
The X-Linked β-thalassemia mental retardation syndrome is a serious recessive disorder resulting from mutations in the ATRX gene, mapping in Xq13.3 and coding for a protein containing the signature motif of the SNF2 family of ATPases. Members of this protein family are thought to act generally as part of large multi-protein complexes and impart on those complexes the ability to utilize the energy of ATP hydrolysis to remodel the structure of chromatin and thereby regulate protein/DNA interactions (11).
This disease is very rare, probably with an incidence of less than 1/100,000 live-born males (11). So far, 173 patients have been reported. Affected males present with severe mental retardation, a typical facial appearance, microcephaly, urogenital anomalies, and an atypical form of β-thalassemia. Pregnancy is usually uneventful, proceeds to term, and, in 90% of cases, birth weight is normal. Affected neonates usually have marked hypotonia and associated feeding difficulties. In early childhood, all milestones are delayed. As the affected individuals grow older, there is often a tendency toward spasticity. Seizures occur in approximately one third of cases. A number of patients have reportedly had jerking movements that are not associated with epileptiform activity on EEG. Brain imaging studies frequently show no abnormality, although mild cerebral atrophy or agenesis of the corpus callosum may be seen. The distinctive facial traits are most readily recognized in early childhood: the frontal hair is often upswept; there is telecanthus, epicanthic folds, midface hypoplasia, and a small, triangular, upturned nose. The upper lip is tented and the lower lip is full and everted, giving the mouth a "carp-like" appearance. The frontal incisors are frequently widely spaced, and the tongue protrudes. Genital abnormalities are seen in 80% of these children. A wide range of relatively mild skeletal abnormalities have been noted. Recurrent vomiting or regurgitation, sometimes treated by fundoplication, is a common finding. The tendency to aspiration is commonly implicated as a cause of death in early childhood. Constipation is common and in some individuals is a major problem to manage. A wide range of cardiac and renal abnormalities also has been noted. There is considerable variation in the hematologic manifestations associated with ATRX mutations (11). A number of families have been identified in which some or all of the affected members have no signs of β-thalassemia; despite lack of these signs, however, diagnosis of β-thalassemia requires only a simple investigation.
Female carriers of ATRX mutations do not have a distinctive phenotype. They are intellectually normal and have no consistent physical manifestations, because they show preferential inactivation of the mutated X-chromosome. Rare cells with HbH inclusions may be found in approximately one fourth of obligate carriers (12). In our case, there were HbH inclusions in 2 of 3 carriers. But recently, there have been some cases of females with ATRX mutations with phenotypical abnormalities. Wada et al. (13) reported a carrier mother who showed non-skewed X-inactivation with moderate mental retardation. Furthermore, Badens et al. (14) described a 4-yr-old girl with typical features of ATR-X syndrome, carrying the recurrent R246C mutation of ATRX. They showed that her pattern of X-inactivation was totally skewed and that her active X chromosome, which harbors the ATRX mutation, was maternally inherited. That was the first report of ATR-X syndrome in a female patient. Fortunately, none of our female carriers had phenotypical abnormalities.
We report the first Korean case of ATR-X syndrome in a 32-month-old boy. Now, we can consider ATR-X syndrome in a young male child with characteristic facial features and neurologic abnormalities of unknown etiology, and get simple screening test for HbH inclusions so that adequate and early management should be started.




 XML Download
XML Download