

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Campomelic dysplasia (CD; OMIM #114290) is a sporadic autosomal dominant disorder that results in skeletal and developmental abnormalities (1, 2). Its reported incidence is about 0.05-0.09 per 10,000 live births (3). Spranger and Maroteaux et al. (4) first described it fully and originally in 1971. CD is a frequently lethal skeletal dysplasia syndrome whose hallmark features include angular bowing and shortening of the long bones with pretibial skin dimpling, hypoplastic scapulae, missing pairs of ribs, a narrow thorax, and bilateral club feet. In addition, hydrocephalus, hydronephrosis, and congenital heart disease (ventriculoseptal defect, atrioseptal defect, aortic stenosis, and/or tetralogy of Fallot) are also present. Patients severely affected by CD usually die from respiratory distress during the neonatal period.
A secondary feature of CD is male-to-female sex reversal, which occurs in about two-thirds of patients with an XY karyotype. Like the sex reversal and the various skeletal symptoms, the bending of the long bones is not an obligatory feature and is absent in about 10% of cases, referred to as acampomelic CD (5). To our knowledge, the present case is the first reported case in Korea of a male with a novel SOX9 frameshift mutation.
CASE DESCRIPTION
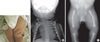
A baby boy was born to a gravida 2 mother with a vertex presentation following a full-term pregnancy in Asan Medical Center's neonatal intensive care unit (NICU) on August 20, 2008. Her first born had been stillborn and was reported to have no anomalies. An ultrasound study at 36 weeks of gestation during this second pregnancy demonstrated shortened long bones, club feet, and a large head. At birth, the one and five minute Apgar scores were 6 and 8, respectively. The boy's birth measurements were as follows: weight 3,272 g (25th percentile), length 46 cm (25th-50th percentile), and head circumference 37 cm (75th-90th percentile). His facial features included a flattened and prominent forehead, a flattened nasal bridge, and short palpebral fissures. Micrognathia and retrognathia were also present, as was a cleft in the soft palate. His ears were set abnormally low. Skeletal deformities included a small thoracic cage, short limbs, pretibial skin dimples on the left thigh (Fig. 1A), anterolateral femoral bowing, and clubbed feet. Laboratory investigations of the blood were normal. A chest radiograph showed a small bell-shaped thoracic cage and 11 pairs of ribs with mild T-L scoliosis and hypoplastic scapulae (Fig. 1B). The pelvic and lower limbs' radiography demonstrated a bowed femur, short fibulae, and no visible talus on either side (Fig. 1C). Pelvic sonography revealed bilateral complete hip dislocations with pseudoacetabular formation. Echocardiography showed a 6.4 mm secundum atrioseptal defect without other malformations. Cerebral ultrasonography on the fifth day showed a small cystic change from both germinal matrix hemorrhages. The brainstem auditory evoked potentials were normal. The patient's karyotype was 46XY with male external genitalia.
To screen for a mutation, we obtained informed consent from the parents for blood sampling. We isolated genomic DNA from peripheral blood using a QuickGene DNA kit (Fujifilm Life Science, Tokyo, Japan). To analyze the SOX9 gene's mutation, we performed PCR using eight sets of primers designed in the intronic flanking region and containing three exons referred to by GenBank accession number NT_010641.15. We performed DNA sequencing using the same primers used in PCR and a BigDye Terminatore V3.1 Cycle Sequencing kit (Applied Biosystems, Foster City, CA, USA). Direct automated sequencing identified a novel frameshift mutation at nucleotide 1372 in exon 3 (Fig. 2). The patient carried both mutant and normal alleles, indicating that the mutation was heterozygous. Both parents declined genetic analysis.
After his discharge from NICU, the patient was hospitalized four times for treatments of pneumonia. He underwent a tracheostomy at the age of three months for severe laryngomalacia and died at the age of four months from progressive respiratory failure.
DISCUSSION
Characteristic features of CD are skeletal hypoplasias and anomalies affecting the face, head, scapulae, spine, pelvis, and upper and lower limbs. The head is macrocephalic with flattened face and nasal bridge, high forehead, low-set ears often with associated deafness, hypertelorism, long philtrum, small mouth, and micrognathia (6-9). The skeletal features are the most prominent characteristics of CD as presented in our case including anterior bowing of the tibia and characteristic pretibial skin dimples. The femurs are also mildly angulated, and talipes equinovarus and dislocation of the hips are usually present. Short fibulae, kyphoscoliosis, brachydactyly and clinodactyly are common. In addition, usually present are flat vertebrae (particularly at the cervical level), hypoplastic scapulae, and a small bell-shaped chest that's often slender with 11 pairs of ribs and a poorly mineralized sternum (7-9). After the critical first year, quality of life tends to improve, although most survived patients are known to be mentally retarded. The oldest reported survivor was a 17-yr-old who had an IQ of 45 (7, 9).
Most cases of CD are caused by heterozygous de novo mutations of the SOX9 gene at chromosome 17q 24.3-q25.1 (5, 10). A growing number of reports describes CD with the chromosome 17 rearrangement breakpoint located some distance from SOX9 (1, 10-12). SOX9 contains a 79-amino acid DNA-binding motif known as the high-mobility-group (HMG) domain, which recognizes typical SOX binding sequences and a second domain essential for its function, a proline/glutamine/serine-rich C-terminal transcription-activation domain (1, 13, 14). SOX9 is a transcription factor that plays a role in the expression of COL2A1, a major collagen gene, and anti-Müllerian hormone, which is secreted from the Sertoli cells for male sex differentiation (15, 16). The mutations cause a loss of either DNA binding or SOX9's transactivation function.
CD is a good model to illustrate how a single transcription factor can control the development of several organs. Both the skeletal dysplasia and the XY sex reversal in CD are caused by mutations in SOX9. All reported mutations in SOX9 can cause CD, and approximately 75% are associated with XY sex reversal; whereas no mutation in SOX9 has been associated with isolated sex reversal (17). The type and location of mutations in SOX9 have demonstrated no correlation with phenotype. Bernard et al. (18) demonstrated that cooperative dimerization of SOX9 was essential for activation of key chondrogenesis genes, but not for male gonadal development, which might explain why CD is not necessarily associated with XY sex reversal as shown in our case.
Four major classes of heterozygous SOX9 mutations cause CD: 1) amino acid substitutions in the HMG domain, 2) truncations or frameshifts that alter the C-terminus, 3) mutations at the splice junction, and 4) chromosomal translocations. All reported missense mutations lie in the HMG domain and affect DNA binding, frameshifts, and splice mutations that truncate SOX9's C-terminus, resulting in the loss of transactivation domains (17, 18). In the present case we identified, a novel frameshift mutation in codon 458 at nucleotide 1372. This mutation altered the transcription activation domain in the C-terminus, leading to a loss of SOX9's function.
To date, two cases of CD have been reported in Korea (19, 20). The first had multiple congenital anomalies (polysplenia, complex heart disease, bilaterally trilobed lungs, and a brain anomaly) and died immediately after delivery. The other was a case in which CD was identified in utero at 30 weeks of gestation whose pregnancy ended in abortion. Neither patient underwent a genetic analysis. Herein we report a novel de novo frameshift mutation (p.Gln458ArgfsX12) in the SOX9 gene of a Korean male with CD.

 XML Download
XML Download