

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The hormonally active form of vitamin D, 1,25-dihydroxyvitamin D3 [1,25(OH)2D3] is an important regulator of bone and mineral metabolism. Its actions are mediated by the vitamin D receptor (VDR) (1, 2).
The hereditary vitamin D resistant rickets (HVDRR) is a rare genetic disorder inherited in a recessive pattern and caused by heterogeneous mutations of VDR gene. The patients with homozygous or compound heterozygous mutations of gene encoding VDR develop the disease (2).
The clinical and laboratory features of HVDRR are rickets, hypocalcemia, hypophosphatemia and increased serum level of 1,25(OH)2D3 due to resistance to VDR (2).
There were a number of reports in a few countries but not in Korea (3). We discovered a case in a three-year old Korean girl. This is the first report of a unique mutation in VDR gene in Korea.
CASE DESCRIPTION
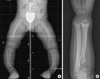
The patient was a 3-yr old Korean girl with curved forearms, bow legs and short stature on September 27, 2007. The height standard deviation score of the patient was -0.73 height SDS compared with the normal growth chart of Korean children that was published in 2007 by the Korean Pediatric Society (4). The radiographic findings of her wrists and ankles were compatible with rickets but she didn't have any type of alopecia (Fig. 1).
Her initial (9/27/2007) serum levels of calcium (7.2 mg/dL, normal range, 8.8-10.8), and phosphorus (3.3 mg/dL, normal range, 3.8-6.5) were decreased, the level of 25-hydroxyvitamin D [25(OH)D] (31.5 ng/mL, normal range, 4.8-52.8) was within normal range, but those of alkaline phosphatase, (1,545 IU/L, normal range, 145-420), 1,25(OH)2D3 (165.0 pg/mL, normal range, 25.1-66.1) and parathyroid hormone (1,197.0 pg/mL, normal range, 9-65) were increased.
After a 3-month treatment of supraphysiologic dose of calcium (100 mg/kg/day) and vitamin D3 (0.2 µg/kg/day), her serum levels of calcium (9.7 mg/dL), phosphorus (5.4 mg/dL) and alkaline phosphatase (510 IU/L) were normalized and her curved forearms were improved. But the bow legs did not show improvement, so she received corrective osteotomies approximately a year later.
Preparation of mRNA and genomic DNA
After obtaining the written informed consent from the patient, her parents and normal children (control group, N = 50) following the Kyung Hee University Hospital at Gangdong Institutional Review Board-informed consent approved protocols, the mRNA and genomic DNA were isolated from their blood samples with Invisorb Spin Blood RNA mini kit [Invitek, Berlin, Germany] and QIAamp DNA Blood mini kit [QIAGEN, Berlin, Germany].
Primer design, PCR and DNA sequencing
The primers for PCR and sequencing reactions of cDNA of VDR are as follows, exon 1a-3 F: 5'-CTG CTT GTC AAA AGG CGG CA-3', exon 1a-3 R: 5'-TTG GGC CGC AGA CTG TCC TT-3', exon 4-6 F: 5'-CTA TTC ACC TGC CCC TTC AA-3', exon 4-6 R: 5'-TGC CAC AGG TCC AGG ACA TG-3', exon 7-9 F: 5'-TGG ACT CGT CCA GCT TCT CCA-3' and exon 7-9 R: 5'-ACT TCG AGC ACA AGG GGC GT-3' (Table 1).
The primers for PCR and sequencing reactions of genomic DNA of VDR are as follows, exon 1a F: 5'-CTG CTT GTC AAA AGG CGG CA-3', exon 1a R: 5'-TTA AAA GAC CCA ACT CCA CC-3', exon 1b F: 5'-TGG AAG GCA AAT AGG AAA CAA-3', exon 1b R: 5'-TAA CTT GCC CAA GGT CAC AA-3', exon 1c F: 5'-CCA ACA AGC CAC TTC CTG TTT-3', exon 1c R: 5'-AGG AAT GAC AGG CAG AGA CTA AA-3', exon 2 F: 5'-AGC TAT GTA GGG CGA ATC AT-3', exon 2 R: 5'-ACA GGC GTA ATG GAA AGA CA-3', exon 3 F: 5'-TGT TGG AGA AAT GGA GAC CA-3', exon 3 R: 5'-GAG TAA CAG GGA GCT GTG AAA AA-3', exon 4 F: 5'-CAC CAT AGC AAA CCC AAT TTT-3', exon 4 R: 5'-TGA TAG GAA TGG CTT TGA GGA-3', exon 5 F: 5'-ATC ATT GCC ATA CTG CTG GA-3', exon 5 R: 5'-TGC ATA GCG CAC AGT ATT TAT G-3', exon 6 F: 5'-TGC CTT ATG CTG CTG AAA GA-3', exon 6 R: 5'-AGG AGA ATC GCT TGA ACC TA-3', exon 7 F: 5'-GCA CAT GAT CAC AGT GTT GAA-3', exon 7 R: 5'-AAC TTG ATG AGG GGC TCA AT-3', exon 8 F: 5'-TCT TTT TCA GCT CCC AGA TTC-3', exon 8 R: 5'-AAC CCT GCT TAT CTA GTT CCT CA-3', exon 9 F: 5'-CGA AGT GTT TGG CAA TGA GA-3' and exon 9 R: 5'-TGC CCG TGA GAA TAT AAC CA-3' (Table 2).
The analysis of the cDNA sequencing was done with 3 primer sets including whole coding regions of cDNA. Their genomic DNA sequencing was done with 11 primer sets including whole coding regions and non-coding regions, including exon/intron splice sites of genomic DNA. Those fragments were sequenced with an ABI 3730XL sequencer (PE Biosystems, Foster city, CA, USA). The sequencing data was analyzed with FinchTV-1.04 sequence aligner (Geospiza, Seattle, WA, USA) and ClustalX-2.0 sequence aligner (Conway Institute UCD, Dublin, Ireland). We compared their sequences with normal ones of VDR gene.
We found a 719 C-to-T transition (Ile146Thr) in exon 4 of the patient and her father's VDR genes, and a 754 C-to-T transition (Arg154Cys) in exon 5 of the patient and her mother's genes (Fig. 2). In this familial study, we concluded that the girl had compound heterozygous mutations in her VDR gene which caused HVDRR.
DISCUSSION
1,25-dihydroxyvitamin D3 or calcitriol [1,25(OH)2D3] is an important regulator of bone and mineral metabolism and its actions are mediated by VDR. The VDR is a member of the nuclear receptor superfamily and belongs to the family of transacting transcriptional regulatory factors and shows sequence similarity to the steroid and thyroid hormone receptors (1, 5).
The heterogeneous mutations of VDR gene cause the hereditary Vitamin D resistant rickets (HVDRR), also known as vitamin D dependent rickets type II (VDDR II) that is a rare recessive genetic disorder (2).
The clinical features of HVDRR are bow legs, curved forearms and specific changes of long bones similar to nutritional rickets, except alopecia in some patients. Also the laboratory findings of HVDRR are hypocalcemia, hypophosphatemia, elevated serum level of alkaline phosphatase and parathyroid hormone as nutritional rickets, except elevated serum 1,25(OH)2D3 level because of end-organ resistance due to mutations in VDR gene (2, 5-7).
The VDR is composed of an N-terminus DNA-binding domain (DBD) and a C-terminus ligand-binding domain (LBD). The VDR binds as a heterodimer with the retinoid X receptor (RXR) to specific vitamin D response elements (VDREs) in target genes via a two-zinc finger DBD. Ligand binding causes a conformational change in the VDR that resets the activation function 2 domain in helix H12 and allows for the recruitment and binding of coactivators that modify chromatin and allow transcription via RNA polymerase II (9-11).
Mutations in the DBD affect VDR-DNA interactions and they result in total loss of VDR transactivation. But, mutations in the LBD cause defects in ligand binding, RXR heterodimerization, and co-activator interaction, and result in partial or total hormone unresponsiveness (9-11).
Alopecia has been associated with mutations that affect DNA binding, RXR heterodimerization and mutations that cause premature stop codons (3, 6, 8, 10, 12-17). But, the cases without alopecia have been usually associated with mutations in ligand binding domain or mutations affecting coactivator protein interactions (9, 11, 18, 19).
More than 40 mutations in the VDR gene have been reported. The types of mutations found in the VDR gene include missense mutations, nonsense mutations, splicing mutations, partial deletions and compound heterozygous mutations (2, 3, 20).
We discovered a three-year old Korean girl who had the typical clinical features of HVDRR including rickets, hypocalcemia, hypophosphatemia, elevated serum calcitriol level and secondary hyperparathyroidism.
The girl and her father were both heterozygous for the 719 C-to-T (I146T) mutation in exon 4, but her father didn't have any symptoms or signs of rickets. Also, the patient and her mother were both heterozygous for 754 C-to-T (R154C) mutation in exon 5 of the VDR gene, her mother didn't have either. This is consistent with previous reports of heterozygous parents being asymptomatic (2).
Because the exon 4 and 5 are in the region of VDR LBD, we think the clinical features of the patient were not as severe and without any type of alopecia, either.
In this familial study, we concluded that each of the patient's parents had a unique heterozygous mutation of the gene and the girl had compound heterozygous mutations in her VDR gene which caused HVDRR. This is the first report of a mutation in the VDR gene in Korea.


 XML Download
XML Download