

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Primary immunodeficiency disease (PID) is a genetically heterogeneous group of disorders that affect distinct components of the innate and adaptive immune system. The cardinal manifestations of PID include frequent respiratory and gastrointestinal tract infections and systemic symptoms, such as fever, weight loss, and failure to thrive. Recent advances in molecular biology have identified more than 120 distinct genes, whose abnormalities account for more than 150 different forms of PID (1). Because of the serious complications associated with PID and the development of diagnostic tools in recent year, clinicians have attempted to determine incidence rates of PID via the collection of clinical data at their home institutions in order to increase diagnosis rates and treatment. However, unreported or undiagnosed subjects remain unaccounted for. Most studies have been performed in European and North American populations; there have been few studies that have examined the incidence of PID among different races and ethnic groups. Published registry data have shown both racial and geographical variation in the prevalence and pattern of PID (2, 3). Within the last 10 yr, PID data from the Middle East and Latin America have been published (4-6). In Asia, the prevalence of PID has been largely unknown, and registries have only been organized in a few countries (7). In Korea, Lee et al. (8) reported the characteristic features of various PID disorders in children over 10-yr at a single center; however, there have only been a few case reports describing PID in Korean adults . This is the first study to investigate the clinical features of PID in Korean adults.
MATERIALS AND METHODS
Patient enrollment, diagnosis, and classification
Fifty-five adult patients who had been diagnosed as having PID at Ajou University Hospital in Korea during a period of 10 yr from January 1998 to January 2009 were enrolled in the study. Individual diagnoses were based on typical clinical features in conjunction with laboratory abnormalities, which were categorized according to WHO criteria (1). We performed laboratory tests on the patients with more than 4 upper and lower respiratory infections within the previous 1 yr, an unusual organism infection, or an unusual duration of treatment. Patients with secondary immunodeficiency diseases such as human immunodeficiency virus (HIV) infection, other viral infections, and chronic systemic steroid or immunosuppressant use were excluded. Medical records were reviewed, and all information, including name, sex, age, family history, previous medications, clinical presentation, types of infections and isolated organisms, allergies, and malignant conversion were recorded. Laboratory analyses were performed using standard techniques and included complete blood counts, platelet counts, examination of peripheral blood smears, erythrocyte sedimentation rates, and complement hemolytic activity (CH50) with C3 and C4. Peripheral blood lymphocyte subsets were analyzed by flow cytometry using a basic panel of T-cell subsets (CD3, CD4, and CD8), B-cells (CD19) and natural killer cells (CD56, CD16). Immunoglobulin G, A, and M were determined using the immunoturbidimetric technique. Reference ranges for normal levels were 916-1,796 mg/dL for IgG; 93-365 mg/dL for IgM; and 40-260 mg/dL for IgA. IgG subclass 1, 2, 3, and 4 concentrations were determined using the single radial immunodiffusion method. Normal ranges for IgG subclass are as follows: 315-855 mg/dL for IgG1; 64-495 mg/dL for IgG2; 23-196 mg/dL for IgG3; and 11-157 mg/dL for IgG4. IgG subclass deficiency was diagnosed in this study if the patient's immunoglobulin levels were 2 standard deviations below the mean of that subclass (9, 10). Genetic testing was performed on only one patient with X-linked agammaglobulinemia. All subjects gave informed consent, as regulated by the Institutional Review Boards of Ajou Medical Center, Suwon, Korea (IRB approval number; AJIRB-CRO-08-215).
RESULTS
Patient characteristics
The mean patient age was 42.5±16.12 yr (ranged from 16 to 76), and most patients were diagnosed as PID during their first visit to the hospital. The patient population was composed of 34 females and 21 males, with a female-to-male ratio of 1.61:1. No patients were reported to have a family history of PID, but related information was limited. Twenty-five patients (25/55, 45.5%) had atopy. Bronchial asthma (33/55, 60.0%) was the most common associated disease. Five patients had only allergic rhinitis and others had autoimmune diseases such as systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and gout. Seven patients (7/55, 12.7%) had both allergies and an autoimmune disease (Table 1).
Frequency and distribution of PID
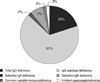
In the present study, 55 adult patients were diagnosed as having PID. All patients were predominantly antibody immunodeficient. In contrast to results in children, none of the patients were identified as having any of the following: combined T- and B-cell immunodeficiency, other well-defined immunodeficiencies, immune dysregulation, and congenital defects in phagocyte number and/or function. Within the predominantly antibody immunodeficiency category, the most common phenotype was IgG subclass deficiency (n=37, 67.3%), followed by total IgG deficiency (n=11, 20.0%), IgM deficiency (n=4, 7.3%), selective IgA deficiency (n=1, 1.8%), common variable immunodeficiency (CVID) (n=1, 1.8%) and X-linked agammaglobulinemia (n=1, 1.8%, Fig. 1). Among patients with IgG subclass deficiency, IgG3 subclass deficiency was the most common (n=17, 47.2%), followed by IgG4 subclass deficiency (n=9, 25.0%). In total, 16.7% of patients with IgG subclass deficiency showed multiple IgG subclass deficiency. Synchronous IgG3 and IgG4 deficiency was most frequently noted. Only two patients showed different disease patterns (IgG1 with IgG4 deficiency and IgG1 with IgG2 deficiency). The mean immunoglobulin values in patients with IgG subclass deficiency were as follows: IgG, 1018.6±380.4 mg/dL; IgA, 226.2±117.7 mg/dL; IgM, 147.5±110.6 mg/dL; IgG1, 628.6±360.7 mg/dL; IgG2, 392.4±189.9 mg/dL; IgG3, 29.5±25.0 mg/dL; and IgG4, 28.9±28.8 mg/dL.
Clinical features
Infectious complications were the major clinical presentation in patients with PID. Several multiple-site infections were noted. The most prevalent infections included respiratory tract infections (including sinusitis), acute pharyngotonsilitis, pneumonia, and bronchitis. The least frequently observed infections were meningitis/encephalitis and genitourinary tract infection. Six patients (10%) had simultaneous multiple site infections such as cellulitis with colitis or urinary tract infection with pneumonia. Respiratory infections were most frequently observed in patients with IgG3 deficiency (Fig. 2A). Bronchial asthma was related to decreased levels of IgG3, IgG4, and multiple IgG subclasses. There was a tendency for patients with autoimmune diseases to have decreased levels of IgG4 (Fig. 2B). Only one patient, who was treated for rheumatoid arthritis, had no history of infection. No mortality was reported in this study, even though some were lost to follow up.
DISCUSSION
In this study, we found that most adults with PID were classified as predominantly antibody deficient, with IgG subclass deficiency as the most common type. This is consistent with results from previous papers, which have reported that IgG subclass deficiency is relatively common (19.4%) in Australia (11). IgG subclass deficiency (26%) and hypogammaglobulinemia (23%) are frequently observed in the USA (12). In contrast, selective IgA deficiency and CVID have been the most frequently observed phenotypes in many countries, based on patients enrolled in a PID registry (4-6, 13, 14). Differences between the results of this study and the distribution of PID in other countries could be due to several factors. First, ethnicity could influence differences in PID distribution. Selective IgA deficiency was found to occur at a ratio of 1:400 to 1:1,000 in European-related populations, but occurred in only 1:14,840 in Japanese blood donors (2, 3). In a Japanese study, selective immunodeficiencies other than IgA accounted for 12.3% of pediatric patients with PID, suggesting it is relatively common in Japanese populations, as compared with western patients (7). Inheritance of the HLA-DR3, HLA-B8, and HLA-A1 haplotype is associated with both common variable immune deficiency and IgA deficiency in European populations. In African American subjects, however, the incidence of HLA-B8 and HLA-A1 antigens was 6 and 7%, respectively, as compared with 16% and 28% for persons of European descent (2, 15). Further, the age of patients in the PID registry ranged from infancy to old age, whereas the patients in this study were in adulthood. Finally, the reported number of PID patients in this study was done in a single center study. Majority of the subjects were those who visited our hospital due to recurrent respiratory infections or unusual infections. Therefore, a national PID registry study based on multicenters will be needed to evaluate the prevalence, distribution, risk factors of the PID in the Korean adult population.
An important finding of this study was that IgG3 subclass deficiency is the most common feature associated with allergic disease, in particular bronchial asthma. These results are consistent with previous studies that have reported IgG subclass deficiency in patients with bronchial asthma or obstructive lung disease (16-18). Other studies have revealed that more than 40% of subjects with IgG3 subclass deficiency have a history of asthma (19) and 41-50% of severe asthmatic children showed IgG3 subclass deficiency (16). Low levels of IgG subclasses have been associated with an increased susceptibility to respiratory infections, especially during primary immune responses to viral protein antigens. In addition, IgG3 is responsible for the primary immune response to Moraxella catarrhalis and the M component of Streptococcus pyogenes, which are the pathogens responsible for exacerbation of upper and lower respiratory infections (20, 21). According to data from the PID registry in Iran, 10 cases of selective IgG subclass deficiency presented with recurrent respiratory infections (4). Recent studies show that 45% of Korean patients with bronchiectasis, who suffered from recurrent respiratory infections, had IgG3 subclass deficiency (21). We hypothesize that IgG3 subclass deficiency is associated with defense mechanisms against viral and atypical pathogen infection, which occur in recurrent upper and lower respiratory infections and the exacerbation of asthma symptoms.
Another finding in this study was that patients diagnosed with SLE or RA had a tendency to present with IgG4 subclass deficiency. Among five patients diagnosed with autoimmune diseases such as SLE or RA, three patients had selective IgG4 deficiency and two patients had combined IgG subclass deficiency (IgG3 and IgG4, IgG1 and IgG4). Epidemiological data have shown that some PID is systematically associated with autoimmune disease. Autoimmune manifestations may occur in PID with variable frequency and include CVID, hyper-IgM syndrome, Good syndrome, and Wiskott-Aldrich syndrome. Rakay, et al. detected the IgG subclass imbalance in 16 patients with connective tissue disease (22). There was also a case report about a 7 yr old boy with IgG2 and IgG4 subclass deficiency who presented with cardiac tamponade due to SLE (23). PID may result in defects in control mechanisms for self-reactive B and T cells, thus favoring the occurrence of autoimmune manifestations. With the exception of one patient, all subjects in this study who were diagnosed as having an autoimmune disease with IgG4 selective deficiency had a history of infections. Although IgG4 represents only a minor portion of total IgG, it may nevertheless be of clinical importance. IgG4 deficient individuals have been reported to suffer from recurrent infections. Heiner suggested that selective IgG4 deficiency might play a role in adult patients with bronchiectasis (24). In pediatric patients, the association between selective IgG4 deficiency and recurrent respiratory infections has been reported. Isolated or combined IgG4 deficiency was most commonly affected, as compared with other isotypes of IgG subclasses in childhood patients (25). In another study, the role of selective IgG4 deficiency in infection-susceptibility was not determined, because it occurred in normal individuals. Approximately 10 to 15% of the general population has IgG4 concentrations below the limit of detection (24). Based on results of previous studies, selective IgG4 deficiency is related to infection and autoimmune diseases. To elucidate the exact role of IgG4, additional clinical and experimental data are needed.
With the development of available genetic testing, more than 120 distinct genes have been identified that may be involved in various forms of PID. Nevertheless, the pathogenesis of IgG subclass deficiency remains largely unknown. In a few cases, homozygous deletions of the corresponding C region genes were documented. But, most cases of IgG subclass deficiency are due to the aberrant regulation of immunoglobulin heavy constant chain gene (IGHG) expression, which is influenced in both a positive and negative manner by various cytokines and B-cell activators (26). Recently, the tumor necrosis factor receptor family member TACI (transmembrane activator and calcium-modulator and cyclophilin ligand interactor) has been found to mediate isotype switching in B cells, which is associated with CVID, selective IgA deficiency, and IgG subclass deficiency (27, 28). Based on the observed infectious complications and B cell isotype switching in patients with IgG subclass deficiency, it is possible that some pathogens (viral or bacterial) could influence isotype switching in B cells, which may cause IgG subclass deficiency in particular patients diagnosed as having bronchial asthma.
The treatment of choice for antibody deficiency is intravenous immunoglobulin (IVIG) replacement therapy (19, 29, 30). Adequate replacement of IVIG has been shown to reduce the incidence of pneumonia and prevent the progression of lung disease in patients with PID. Similar effects of IVIG were noted in the patients with IgG subclass deficiency. Subjects in this study were administrated IVIG monthly; IVIG significantly improved quality of life, decreased the number of infections and the need for antibiotics, and improved IgG subclass levels (30). When IVIG was used as a prophylaxis, significant protection against infection was noted (19).
In conclusion, this is the first study of adult PID from a single tertiary medical center in Korea. IgG subclass deficiency was the most common disease in adult patients and IgG3 and IgG4 were commonly affected. The main clinical presentations included infectious complications, especially respiratory infections. Therefore, IgG subclass deficiency should be considered in adult patients with recurrent upper and lower respiratory infections, particularly in those with asthma or autoimmune diseases.


 XML Download
XML Download