

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multicystic dysplastic kidney (MCDK) is a relatively common developmental anomaly in infants and children, and its overall prognosis is good in older children. Although MCDK is grossly "cystic" in appearance, it is not one of the inherited renal cystic diseases, but a kind of renal dysplasia, in which cystic elements are found along with immature, undifferentiated, primitive tissue (1). In contrast, a malignant rhabdoid tumor of the kidney (MRTK) is one of the most lethal neoplasms of early life, and the mortality rate exceeds 80% (2). MRTK usually arises from perihilar renal parenchyma and infiltrates into the medulla, renal sinus, and collecting system. The size of the tumor is usually greater than 9 cm in diameter. The recurrence rate is high, and the tumor tends to metastasize to the lung, liver, and brain (2, 3). There have been some reports concerning MRTK and MCDK presenting individually and MCDK presenting with Wilms tumor (4). However, a combined case of these two diseases has not been reported to date. Therefore, the authors present a case of MRTK combined with MCDK in a 5-yr-old girl with a literature review.
CASE REPORT
A 5-yr-old girl who was previously diagnosed with MCDK at birth by abdominal sonography presented with a huge palpable mass on the right side of her abdomen. No symptoms were noted until a huge mass was palpated two weeks prior to presentation. Abdominal sonography and computed tomography (CT) were performed, and they revealed an increased cystic lesion extensively replacing the right kidney and a newly-developed amorphous portion in the cystic lesion, compared to the previous radiologic findings at birth (Fig. 1). These findings suggested an infectious change of MCDK. Right nephrectomy was performed. On the laboratory test of peripheral blood, the level of creatinine was within normal range.
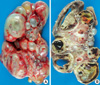
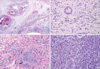
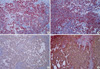
On gross examination, the right kidney was extensively replaced by numerous dilated cysts (Fig. 2A). The cut surface showed numerous cystic and semisolid masses in the cortex and medulla (Fig. 2B). Microscopically, the individual tumor cells had diffusely infiltrated the kidney parenchyma. Multicystic changes with diffuse necrosis were also noted (Fig. 3A). Some of the neoplastic cells had eccentric nuclei and large, round, eosinophilic cytoplasmic inclusions. In the remaining parenchyma, there were immature glomeruli and metaplastic cartilage (Fig. 3B, C). The tumor cells were non-cohesive, large, round-to-polygonal cells with vesicular nuclei and prominent nucleoli (Fig. 3D). The immunohistochemical stains of the rhabdoid tumor cells were positive for cytokeratin, vimentin, INI1, and epithelial membrane antigen and negative for desmin and smooth muscle actin (Fig. 4). Ultrastructurally, the tumor cells had poorly formed intercellular junctions, a fair amount of mitochondria, and were filled with lipid droplets (not shown).
DISCUSSION
Generally, kidney abnormalities are classified by quantity, location, morphology, differentiation, and genetic disorder. Among them, MCDK is a common congenital disease seen in infants (5). One in 4,300 live births is estimated to have unilateral MCDK (6), and few occur in syndromes of multiple malformations. Mainly, MCDK occurs unilaterally and is associated with urinary obstruction (5). In contrast, MRTK is a rare malignant tumor of the kidney, and it accounts for about 2-3% of kidney tumors (3). MRTK also tends to occur in young infants and is one of the most lethal neoplasms in early life (7). Before 1981, investigators thought MRTK was a kind of Wilms tumor; however, Haas et al. (2) found otherwise. They reviewed 111 cases of rhabdoid tumors of the kidney by analyzing microscopic findings, immunohistochemical stains, and electron microscopic findings of the cases. Unlike with a Wilms tumor, they found that some of the tumor cells originated from primitive cells. These tumors showed different histological characteristics from that of a Wilms tumor. Therefore, they called the disease "MRTK". Some investigators suggested that MRTK might originate from widely-distributed precursor cells or neuroepithelial cells (7). There has not been a report about a case of MCDK presenting concurrently with MRTK with an examination of the cause of their coexistence and their pathophysiology to date.
Embryologically, dysplastic kidney disease, which is induced by a metanephron abnormality, is irreversible, and the severity of disease and affected areas are varied (5). MCDK is a common disease in infants, yet the pathophysiology of MCDK is not well understood. One hypothesis is that MCDK results from an abnormal induction of the metanephric blastema by the ureteric bud (5). Embryologically, MCDK may result from abnormal renal morphogenesis, likely due to abnormalities of developmentally expressed genes (1). Until now, PAX2, BCL2, and galectin-3 genes were thought to be related with the occurrence of MCDK, and chromosome t(6:19)(p21:q13.1) abnormality-induced cytogenetic change (8). Those genes are associated with oncogenesis and their high level of expression can accelerate the proliferation of dysplastic cysts, which causes some patients' multicystic dysplastic kidney to continue to propagate (8, 9). On the other hand, about 15% of MRTKs are characteristically combined with non-rhabdoid tumor of primary or metastatic CNS disease. They arise embryologically from the stromal cell, mesenchymal cell, neuroectodermal cell, or germ cell. It is reported that genetically 90% of rhabdoid tumors have a 22q11.2 chromosomal translocation, and a mutation or deletion of SMARCB1, HSNF5/IML, and DDT1 genes (10). According the theory of pathogenesis, the two kinds of disease are probably related to stem cell mutation. However, the common chromosomal abnormality was not confirmed until now.
Distinctions should be made among MCDK, Wilms tumor, MRTK with a cystic pattern, and other diseases affecting the kidney. The pathologic findings in this case revealed a typical MCDK that coincided with embryonic tubules, glomeruloid structures, and immature cartilage. More importantly, condensing mesenchyma surrounded the central ureteric bud. These findings support the diagnosis of MCDK in this case. In addition, immunohistochemical stains were positive for cytokeratin, epithelial membrane antigen, and vimentin, but negative for smooth muscle actin. Therefore, it was diagnosed as MCDK coexistent with MRTK (11). By electron microscopy, tumor cells of usual cases of MRTK form sheetlike patterns, have poorly formed intercellular junctions, abundant mitochondria, and lipid droplets in the cytoplasm, especially prominent aggregates of filaments. However, in this case, the cytoplasm did not have prominent aggregates of filaments. Therefore, the diagnosis of MRTK should be given only after thorough investigations by light microscopy, immunohistochemical stains, and electron microscopy.
Most MCDKs have a good prognosis. They generally regress within the first two years of life. Age is a highly significant prognostic factor, and the younger the patient, the better the prognosis. Subjects older than five years are less responsive to chemotherapy, suggesting a poor prognosis (12). Thus, conservative management is the recommended treatment option. However, when the disease is combined with hypertension or a malignant tumor, the patient is more likely to have a poor prognosis (13). After being diagnosed with MCDK, the patient needs regular follow-up to ensure that the cystic tumor is regressing or to uncover any evidence that a new neoplasm is developing. Until now, there have already been reports of MCDK simultaneously occurring with renal cell carcinoma, mesothelioma, and Wilms tumor (14). The prognosis of MRTK is related to the stage, lymph node metastasis, sex, nuclear diameter, and other factors. In this case, because the patient was 5 yr old, the tumor was stage I, and the cells had a medium nuclear size (2-4 µm), according to the MRTK prognosis markers, the patient might have belonged to the favorable prognostic group. However, MCDK combined with MRTK has not been reported to date. It is difficult to evaluate the patient's prognosis when there are two different diseases present at the same time. However, the patient is in good condition after surgery. Hence, a prognosis should be related to the more serious disease, if two diseases occur simultaneously.
In conclusion, the authors believe that this case, MCDK combined with MRTK found in an infant, is the first of its kind to be reported, and we consider that this occurrence can become a new disease entity in infantile renal disease in the future. According to the literature, MCDK and MRTK are different embryologically, epidemiologically, and genetically. For this case, it may be possible to suggest how this coexistence developed. The first possible mechanism is that the two diseases may have occurred simultaneously. In contrast, each of them may have developed independently. Finally, early development of one could have affected the course of the other. By reviewing more cases and studies, pathogenetic and prognostic evidence of MCDK combined with MRTK may be identified in the future.

 XML Download
XML Download