

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
With advances in surgical technique, improvements in perioperative care, and better criteria for patient selection, the mortality rates during liver resection and transplantation have decreased to less than 5%. However, ischemia and reperfusion (I/R) injury during liver surgery is an inevitable, unsolved problem that often leads to fatal, irreversible postoperative liver failure.
Assessment of liver function is an important clinical issue in liver surgery. For the liver, many biochemical values and parameters including energy charge level, adenosine triphosphate (ATP) in the liver tissue, and ketone body ratio have been used to measure liver function and functional reserve (1-4). However, these conventional methods are limited in assessing liver function during surgery: time is required for sample acquisition and analysis, and sometimes the results are acquired too late to take steps toward preventing irreversible postoperative liver failure.
Because there is no direct, practical method for assessing liver function or cell viability during liver surgery in a real-time manner (like an ECG in heart disease), we based this study on the fact that ischemia causes biochemical and physiological changes in living tissue, which influence its bioelectrical impedance (BEI) (5). This study was undertaken to work toward a real-time I/R monitoring system using BEI quantification and liver cell viability during liver surgery.
MATERIALS AND METHODS
Animals
Male Sprague-Dawley rats (weighing 200-250 g) purchased from Hyo-chang Science (Daegu, Korea) were used for this study. The animals were fed rat chow and given free access to water. The care, maintenance, and treatment of the animals in this experiment followed the protocol 2008-11 approved by the Institutional Animal Care and Use Committee of Yeungnam University College of Medicine. Rats were divided into ischemia group (n=5, 120 min of ischemia) and I/R group (n=5 for each time point, 24 hr of reperfusion after 30, 60, and 120 min of ischemia).
Liver ischemia and bioelectrical impedance measurement
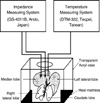
Under anesthesia with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine® (10 mg/kg), a midline laparotomy was performed. A model of 70% partial hepatic ischemia proposed by Camargo et al. (6) was used in all experiments. In order to implement 70% ischemia, hepatoduodenal ligament was dissected and a microvascular clamp was placed on the portal pedicle to median and left lobe of liver. Reperfusion was performed by removing the microvascular clamp and the abdomen was closed. The animals were housed in separate cages maintained at room temperature, and were allowed to recover with free access to rat chow and water ad libitum. After 24 hr of reperfusion, the rats were anesthetized with ketamine and xylazine again, and the liver tissue was collected immediately for further analysis. BEI was measured at various frequencies (0.12, 1.2, 12, 62.5, and 100 KHz) with a multi-frequency LCR meter (GS-4311B, Ando, Japan) every 10 min. Two platinum electrodes (length 25 mm, diameter 0.45 mm) were inserted into the median lobe of the liver. The distance from each electrode was 10 mm. The electrodes were coated with Teflon to maintain adequate insulation except the tip (2 mm length). A heat mattress and transparent acryl case were used to maintain the body temperature and humidity of each rat (Fig. 1).
Measurement of ATP
Approximately 100 mg of fresh median lobe liver tissue was excised 30 min interval during ischemia and after 24 hr of reperfusion. All specimens were immediately placed in ice-cold 10% HClO4 buffer and homogenized with an Ultraturax T-25 (Janke & Kunkel, Staufen, Germany). The ATP content of the liver tissue was measured according to the method of Khan (7). The homogenate was centrifuged at 4,500 rpm for 10 min at 4℃. The supernatant was neutralized with 200 µL of 2.5 M/L KOH, and the precipitate was removed by centrifugation at 4,500 rpm for 5 min at 4℃. The supernatant was mixed with 10 mM/L Tris-acetate buffer and was kept on ice. The ATP relative luminescence unit (RLU) was measured with ATP Assay Kit (ENLITEN® ATP Assay System Bioluminescence Detection Kit for ATP, Promega, Madison, WI, USA) by using a luminometer (TD-20/20, Turner Biosystems, Sunnyvale, CA, USA).
Measurement of palmitic acid oxidation rate in the liver
Approximately 500 mg of fresh tissue from the median lobe was excised 30 min interval during ischemia and after 24 hr of reperfusion. All specimens were immediately placed into SETH buffer (250 mM sucrose, 1 mM EDTA, 10 mM Tris-HCl, 5 mM ATP, pH 7.4). The tissue samples were minced, and SETH buffer was added to yield a 20-fold diluted homogenate. The liver suspension was transferred to 3 mL glass homogenization tube and homogenized with a motor-driven teflon pestle. The liver tissue fatty acid oxidation rate was measured according to the method of Kim et al. (8). A 160 µL of reaction mixture (100 mM sucrose, 10 mM Tris-HCl, 5 mM potassium phosphate, 80 mM potassium chloride, 1 mM magnesium chiloride, 1 mM L-carnitine, 0.1 mM malate, 2 mM ATP, 10 mM CoA, 100 mM palmitate, 1 mM dithiothreitol, 0.1 mM EDTA, and 0.5% bovine serum albumin) was added to 40 µL quantity of a 20-fold diluted homogenate. After incubating for 60 min at 30℃, 100 µL of perchloric acid was injected to stop the reaction. Sodium hydroxide (200 µL) was used to trap the CO2 produced during the 60 min incubation. After mixed with 5 mL cocktail, a liquid scintillation counter was used to measure the palmitic acid metabolic rate of the liver tissue homogenates.
Histology
The median lobe liver tissue was excised 30 min interval during ischemia and after 24 hr of reperfusion. All excised liver tissues were fixed in 10% formalin, dehydrated, and embedded in paraffin. Sections (4 µm) were stained with both hematoxylin and eosin (H&E) and the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay (ApopTag Peroxidase In Situ Apoptosis Detection Kit, Chemicon, Temecula, CA, USA).
Statistical analysis
All data are expressed as mean±SE. Data were analyzed using SPSS 12.0 statistical package. Mean values were compared using ANOVA with repeated measures and Student's t-test to evaluate of significant differences. A P-value of less than 0.05 was considered statistically significant.
RESULTS
Experiment I. Ischemia group
The BEI changed significantly during ischemia at low frequency compared to high frequency. The BEI at 0.12 KHz increased gradually during 60 min of ischemia and had a tendency to plateau thereafter (Fig. 2).
The ATP content decreased to below 20% of the baseline level after 30, 60, and 120 min of ischemia, and there was no significant difference between 60 and 120 min of ischemia. The ATP (µM/g wet liver tissue) contents were 2.5±0.2, 0.6±0.1, 0.3±0.1, and 0.2±0.1, at 0, 30, 60, and 120 min of ischemia, respectively (Fig. 3).
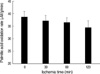
The palmitic acid oxidation rates (nM/g/min) were 38.7±2.1, 37.0±2.2, 36.6±1.8, and 34.6±2.6 at 0, 30, 60, and 120 min of ischemia, respectively. There were no significant differences among 30, 60, and 120 min of ischemia (Fig. 4).
There were no significant differences in histological examination based on H&E and TUNEL techniques throughout the isochmia (Fig. 5).
Experiment II. Ischemia-reperfusion group
The BEI was significantly increased after 30, 60, and 120 min of ischemia. BEI at 60 and 120 min of ischemia were higher than in 30 min of ischemia. BEI was recovered to near baseline level, but lesser according to the duration of ischemia after 60 min of reperfusion (Fig. 6).
The ATP content decreased significantly after 24 hr of reperfusion following 30, 60, and 120 min of ischemia, compared to that of normal liver (non-ischemia). The ATP (µM/g wet liver tissue) contents were 2.7±0.1, 1.3±0.2, 0.9±0.1, and 0.8±0.1 at 24 hr of reperfusion after 0, 30, 60, and 120 min of ischemia, respectively. There was no significant difference between 60 and 120 min of ischemia (Fig. 7).
The palmitic acid oxidation rate was significantly decreased after 24 hr of reperfusion following 30, 60, and 120 min of ischemia, compared with that of normal liver (non-ischemia). The palmitic acid oxidation rates (nM/g/min) were 36.6±2.6, 33.3±2.7, 28.5±2.0, and 27.1±3.3 after 24 hr of reperfusion following 0, 30, 60, and 120 min of ischemia, respectively. There was no significant difference between 60 and 120 min of ischemia (Fig. 8).
In histological examination by H&E and TUNEL stating, hepatic necrosis and apoptosis were observed in 2.0±0.8, 9.4±1.1, and 12.0±1.7% of the whole liver, after 30, 60, and 120 min of ischemia and 24 hr of reperfusion, respectively. The amount of hepatic necrosis after 60 or 120 min of ischemia was significantly higher than that seen after 30 min of ischemia, but there was no significant difference between 60 and 120 min of ischemia (Fig. 9).
DISCUSSION
The liver is known to be more susceptible to I/R injury than any other organ except the brain. However, I/R injury is an unavoidable problem during liver surgery, and it often results in fatal complications leading to irreversible liver failure and death after surgery.
Before liver resection is performed, assessment of hepatic functional reserve should be undertaken. Because postoperative liver failure is difficult to treat, preoperative assessment of liver function and postoperative assessment of remaining functional liver mass and its reserve are of paramount importance to minimize surgical risk. There are varieties of hepatic functional reserve tests, such as the traditional Child Pugh classification, the clearance of indocyanine green, the redox-tolerance index, the C-aminopyrine breath test, and the galactose elimination test (9-12). However, these tests are not direct or practical for predicting liver cell damage in a real-time manner during liver surgery.
The investigation of the passive electrical properties in biological tissues to see pathologic changes has been important issue since the end of 19th century. One of the most common tools to see tissue electrical properties has been BEI. Nowadays, BEI monitoring is an emerging tool for biomedical research and medical practice. It is used for measuring the cardiac output, tissue blood flow, assessment of body composition, radiography tomography etc, based on difference in the electrical properties of each biologic tissue (13). Regarding ischemia and BEI, Konishi et al. (13) reported the possibility of estimating changes in liver tissue due to ischemia with BEI technique. It has been also established that ischemia causes biochemical and physiological changes in tissue, which influence its own BEI (5). We did this study based on the assumption that biochemical and physiologic change in living liver tissue during I/R can be monitored with BEI in a real-time manner.
The electrical properties of biological tissues show frequency dependence (13). As shown in Fig. 2, the BEI changed significantly during ischemia at low frequency versus high frequency. For the measurement of BEI, we inserted two platinum electrodes in the median lobe of the liver. Because of the small cell size compared to the size of the electrodes, we assumed the tips of the electrodes would be in the extracellular space. At low frequency, the current does not pass through the cell membrane (14), so the impedance is responsible for the resistance of the extracellular fluid.
Temperature can also influence change of BEI. We tried to maintain temperature as high as possible using heat mattress and acryl case. Actual temperature of liver measured with DTM 322 (Tecpel, Taiwan) were 36.7±3 and 32.1±3℃ at 0 and 120 min of ischemia. But conductivity temperature coefficiency is where between 1%/℃ and 2%/℃ (15, 16). Konishi et al. (13) also concluded that even though there was negative correlation between temperature and extracellular resistance, main cause of BEI change in extracted liver is ischemia. In our experiment, BEI change during I/R was beyond the change expected with conductivity temperature coefficiency.
During ischemia, the oxygen and nutrient supplies are stopped, resulting in a loss of mitochondrial oxidative phosphorylation and a decrease in ATP generation. The depletion of cellular ATP stores induces alterations in transmembrane ion transport through inhibition of the ATP-dependent Na+/K+ ATPase. This leads to sodium and chloride influx changes, intracellular sodium accumulation, and eventually in cell swelling (17, 18). Although we did not definitively clarify the reason for BEI changes, we hypothesized that changes in the ion concentration in the extracellular fluid caused the BEI changes.
The BEI (0.12 KHz) increased gradually during the 60 min of ischemia and had a tendency to plateau thereafter. After 60 min of ischemia, the ATP content decreased to below 20% of the normal level, but there was no significant difference between 60 and 120 min of ischemia. We used palmitic acid as standard free fatty acid to see cell viability during ischemia and reperfusion in rat liver. Hepatocytes can generate ATP from oxidations of free fatty acids and glucose. In other words, dead cells do not produce ATP, and damaged cells produce much lesser ATP than normal cells. In this point, free fatty acids or glucose oxidation rate indicates cell viability or cellular function (19). There were no significant changes in the palmitic acid oxidation rate or histological examination (H&E and TUNEL staining) during 120 min of ischemia. Even though ischemia reduced the ATP content to below 20% of the normal level, the liver cells still had the ability to metabolize palmitic acid. Based on these results, we believe the liver has relatively good tolerance to ischemia.
ATP depletion is an important contributing factor that increases liver cell injury after reperfusion. The hepatic ATP concentration has been used as an indicator of liver function (20). BEI was significantly increased after 30, 60, and 120 min of ischemia. After 60 min of reperfusion, BEI was recovered according to duration of ischemia. The ATP content decreased to below 50% of normal in all the reperfusion groups. During a subsequent reperfusion phase, oxidative phosphorylation resumes, and ATP is used for membrane repair and ion pumping (21). After 24 hr of reperfusion in our study, the palmitic acid oxidation rates had decreased significantly compared with normal liver. These results suggest to us that the liver tolerates ischemia relatively well, but is susceptible to reperfusion injury.
Regarding hepatic ischemia without portal venous drainage, the upper time limit is reported to be 60 min (22), but some investigators have reported a limit in excess of 90 min in normal liver (23). In this study, the 70% partial hepatic ischemia model was used, because survival was poor after 90 min of total ischemia. Even though there was no significant change in histology at 30, 60, and 120 min of ischemia, hepatic necrosis and apoptosis were observed in 2.0±0.8, 9.4±1.1, and 12.0±1.7% of the whole liver after 24 hr reperfusion following 30, 60, and 120 min of ischemia, respectively. Based on these results, we noted that liver damage after reperfusion depended on the duration of ischemia.
In conclusions, BEI may be a good tool to monitor I/R during liver surgery. The liver tolerates ischemia relatively well, but after reperfusion, liver cells may be damaged depending on the duration of ischemia. This study might be helpful as a guideline for predicting liver damage after I/R in both clinical practice and basic research.







 XML Download
XML Download