

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatitis B virus (HBV) infects more than 350 million people worldwide and is one of the major causes of chronic liver disease in Asia-Pacific region. Chronic HBV carriers manifest variable stages of liver inflammation and fibrosis in both clinical and pathologic aspects. The most serious outcome of those is cirrhosis that is not only a major cause of liver related death but also a main risk factor of hepatocellular carcinoma (HCC) in chronic HBV carriers. Therefore, the objectives of many basic researches and antiviral drugs for HBV were to prevent or retard the progression of cirrhosis. Among chronic HBV carriers, there are individual differences in the progression of cirrhosis. Even some of them never progress to cirrhosis. However, very few studies have assessed the progression rate of fibrosis or cirrhosis in chronic HBV carriers because of complex pathogenesis and the requirement of long term observation.
Several individual gene differences influence the progression rate of fibrosis. One of those is a cytokine gene polymorphism that influences the production or the activation of cytokines, linked to inflammation and fibrosis.
Transforming growth factor (TGF)-β is a multifunctional cytokine that regulates variety of cellular processes including proliferation, differentiation, apoptosis, angiogenesis and wound healing related to produce extracellular matrix. There are three isoforms of TGF-β (TGF-β1, -β2, and -β3) that are encoded by distinct genes, but have similar biologic actions (1). Of these, TGF-β1 is a major cytokine that contributes to liver fibrosis via activating hepatic stellate cells and increasing the synthesis of extracellular matrix (2).
Seven genetic polymorphisms of TGF-β1 have been identified: 3 in the upstream region of the gene at positions -988, -800, and -509; 1 in a nontranslated region at position +72; 2 in the signal peptide sequence region at codon 10 (position +869 C or T) and 25 (position +915 G or C); and 1 in the protein coding region at codon 629 (3). TGF-β1 gene polymorphisms at codon 10 and 25 affect the amounts of TGF-β1 production in vivo and in vitro. In the lungs, leucine homozygous genotype (L/L, +869 T/T) at codon 10 was associated with increased serum level of TGF-β1 and lung fibrosis (4). Arginine homozygous genotype (+915 G/G) at codon 25 was associated with in vitro increased leukocyte production of TGF-β1 and lung fibrosis (4). In the liver, arginine homozygous genotype at codon 25 was associated with liver fibrosis (5). However, genetic polymorphism at codon 10 showed conflicting results as to which genotype was more fibrogenic in the liver (5-9). These conflicting results could be attributed to the complex pathogenesis and various factors that contribute to liver fibrosis.
There has been no genetic polymorphism at codon 25 in Koreans (10). Present study investigated whether the genetic polymorphism at codon 10 is associated with the development of cirrhosis in chronic HBV carriers.
MATERIALS AND METHODS
Patients
Two hundred and twenty Korean patients with cirrhosis (including HCC with underlying cirrhosis), who had hepatitis B surface antigen (HBsAg, RIA, Abbott Laboratory, Chicago, IL, USA) for over 6 months and no hepatitis C virus antibody (anti-HCV, RIA, General Biologicals Corp, Hsin Chu, Taiwan), were admitted to Gil Hospital, Incheon, Korea from January 2001 to January 2005. Among those, one hundred and twenty-one patients who had alcohol intake of less than 20 g/day and were over 50 yr old were assigned to liver cirrhosis (LC) group. Eighteen patients in LC group were admitted due to non liver related symptoms and diseases. The other 103 patients visited hospital due to liver related symptoms. Diagnosis of cirrhosis was based on at least 2 of the followings: 1) gastroesophageal varices on endoscopy, 2) cirrhotic surface or regenerating nodules of liver and 3) splenomegaly by radiologic images (ultrasonography or computed tomography).
Eighty-four Korean chronic hepatitis B patients, who had HBsAg for over 6 months, no anti-HCV, no clinical signs of cirrhosis (above criteria), alcohol intake of less than 20 g/day and were over 50 yr old, were admitted to the same hospital during the same periods. Among these, 9 patients were excluded because they had history of antiviral treatment for over one year. Eighteen patients were also excluded because their platelet counts were less than 150,000/µL. Therefore, 57 chronic hepatitis B patients were enrolled and assigned to chronic hepatitis (CH) group. In CH group, 17 patients were admitted due to liver related symptoms. Twenty-five patients visited the hospital due to non liver related diseases and symptoms. Fifteen patients were incidentally diagnosed as chronic hepatitis B by National health examination.
Determination of clinical and laboratory parameters
All patients' laboratory data including hepatitis B e antigen (HBeAg, RIA, Dainabot Co., Tokyo, Japan) and the level of HBV DNA (Diagene Inc., Basel, Switzerland) were collected within 6 months at enrollment. However, if patients were using antiviral agents, those data before antiviral treatment were taken for analysis. Whole blood samples were collected and stored at -70℃ refrigerator for genetic assay.
All patients gave informed consents for blood sampling and using personal data. The study protocol was approved by institutional review board of Gil Hospital, Incheon, Korea and in accordance with the Helsinki Declaration.
Genetic polymorphism at codon 10 in TGF-β1
Genomic DNAs were extracted from peripheral blood leukocytes using proteinase K and phenol/chloroform. Polymerase chain reaction (PCR) was performed by using a primer set (sense 5'-TGT TCG CGC TCT CGG CAG T-3', antisense 5'-TCA CCA GCT CCA TGT CGA TA-3') that amplified the DNA fragment including codon 10. PCR mixture consisted of MgCl2 1.5 mM/L, KCl 50 mM/L, Tris-HCl 10 mM/L, dNTP 200 M/L, primer 0.5 µM/L, Taq polymerase 1 U (Bioneer, Daejeon, Korea), and genomic DNA 50 ng. After initial denaturation at 94℃ for 5 min, thermocycling consisted of denaturation at 94℃ for 30 sec, annealing at 60℃ for 30 sec, extension at 72℃ for 30 sec for 35 cycles, and followed by a final extension at 72℃ for 5 min.
For the single stranded conformational polymorphism (SSCP) analysis, 2 µL of the PCR product were mixed with 8 µL of denaturing solution containing formamide, 250 mM NaOH, 25 mM EDTA, and 0.05% bromophenol blue. Then the DNA samples were denatured at 94℃ for 5 min, dipped in ice and loaded onto a 13.5% polyacrylamide (acrylamide to bisacrylamide ratio of 29:1) gel. Single-stranded DNA migrated through the gel at 40 W power for 6 hr in a 4℃ cold room and visualized by silver staining.
To define the genotyping results, selected above PCR-amplified DNA samples (n=10, respectively, for each SSCP patterns) were examined by DNA sequencing.
Statistical analysis
Continuous variables were expressed as mean æstandard deviation. The Student t-test and chi-square-test were used to compare variables between LC and CH groups. The consistency of genotype frequencies with Hardy-Weinberg equilibrium was checked. The multivariate logistic regression analysis was used for identifying independent risk factors of the development of cirrhosis. A P value less than 0.1 in univariate analysis and 0.05 in multivariate analysis were considered statistically significant. The SPSS version 10.0 (SPSS, Chicago, IL, USA) was used for statistical analysis.
RESULTS
Clinical characteristics of LC and CH groups
As shown in Table 1, mean age and sex ratio were not different between LC and CH groups. The LC group showed abnormal laboratory features of deteriorated liver functions and portal hypertension such as low albumin level, high total bilirubin level, low platelet count, and prolonged prothrombin time. In contrast, the CH group had minor laboratory abnormalities in those features. HCC was present in 70 patients in LC group, but absent in CH group. The HBeAg positivity and the detection rate of HBV DNA by hybridization capture method were higher in LC than in CH group (P=0.055 and P=0.003, respectively).
Distribution of genotypes at codon 10 in TGF-β1
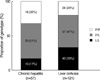
The SSCP results according to TGF-β1 gene polymorphism at codon 10 showed 3 different patterns (Fig. 1). The 2 bands, 3 bands, and 1 band represented proline homozygous (P/P) genotype, heterozygous (P/L) genotype, and leucine homozygous (L/L) genotype, respectively. These genotypes were confirmed by direct DNA sequencing as previously mentioned. The distributions of genotype at codon 10 in LC and CH groups are shown in Fig. 2. In CH group, the proportions of P/P, P/L, and L/L genotypes were 32%, 51%, and 17%, respectively. In LC group, the proportions of P/P, P/L, and L/L genotypes were 20%, 47%, and 33%, respectively. The proportion of P/L genotype was similar between both study groups. The proportion of P/P genotype was higher than that of L/L genotype in CH group, whereas the proportion of L/L genotype was higher than that of P/P genotype in LC group. When the proportions of P/P and L/L genotypes were compared between both study groups, the proportion of L/L genotype was higher in LC than in CH group (P=0.017). Conversely, the proportion of P/P genotype was higher in CH than in LC group (P=0.017). If cirrhotic patients without HCC (n=51) were selected in LC group, the proportions of P/P, P/L, and LL genotypes were 18%, 41%, and 41%, respectively. The proportion of L/L genotype was higher in cirrhotic patients without HCC than in CH group and the proportion of P/P genotype was higher in CH group than in cirrhotic patients without HCC (P=0.008).
Evaluation of the risk factors for the development of cirrhosis
In univariate analysis, the HBeAg positivity, detection rate of HBV DNA, and distribution of genotype were significantly different between both study groups. Therefore, multivariate analysis for the risk factors of the development of cirrhosis was performed with the above three significant variables. Multivariate logistic regression analysis revealed that detectable HBV DNA and L/L genotype were the only risk factors for cirrhosis (Table 2). In comparing with undetectable HBV DNA, the detectable HBV DNA showed an odds ratio of 3.037 (95% confidence interval [CI]; 1.504 to 6.133, P=0.002). In comparing with P/P genotype at codon 10, the L/L genotype had an odds ratio of 3.408 (95% CI; 1.279 to 9.085, P=0.014).
DISCUSSION
There are conflicting results in literature as to which genotype at codon 10 is associated with more production of TGF-β1 or fibrosis than others. The L/L genotype was associated with increased serum level of TGF-β1 and lung fibrosis (4). However, other report suggested that the P/P genotype was associated with increased serum level of TGF-β1 (11) and liver fibrosis (8, 9). On the other hand, some reports found no association between genotype at codon 10 and liver fibrosis (5-7). These contradictory results may be attributed to the heterogeneity of populations, races, diseases, and other confounding factors.
Present study indicated that patients with L/L genotype at codon 10 in TGF-β1 had higher incidence of cirrhosis than those with P/P genotype. In other words, patients with P/P genotype at codon 10 had lower risk of progressing to cirrhosis than those with L/L genotype. This association was significant after correcting for potential confounding variables (age, sex, HBeAg, and HBV DNA) that might independently affect the development of cirrhosis.
In our previous report, we investigated the distribution of genotype at codon 10 in 119 healthy controls that had not HBsAg or anti-HCV. The proportions of P/P, P/L, and LL genotypes were 26%, 45%, and 29%, respectively (12). The proportion of P/P was similar to that of L/L genotype. In other reports from Japan, China, and Korea about distribution of genotype at codon 10 in general population, the proportion of P/P was similar to that of L/L genotype (6, 9, 13). However, the P/P genotype was presented more frequently in CH group than in healthy controls or LC group. In otherwise, L/L genotype was presented more frequently in LC group than in healthy controls or CH group.
The production and activity of TGF-β1 are regulated by complex posttranscriptional processes, including mRNA stabilization, the assembly and activation of the latent TGF-β1 complex, and the modulation of receptor expression (14). The genetic polymorphism at codon 10 presents within the signal sequence that is coding amino acids and conforms the prepro-TGF-β1 with pro-TGF-β1 protein (15). Because most of general genetic polymorphisms are located in the noncoding regions of gene, it is very difficult to prove whether those have direct impact on the phenotype of diseases (16). However, the genetic polymorphisms in coding region alter the function or structure of encoded proteins. Those alterations can directly influence the phenotype of diseases. Therefore we assumed that the genetic polymorphism at codon 10 may affect the production or activity of TGF-β1, because it presents within the coding region.
The function of a signal sequence is translocation of newly synthesized proteins across the membrane of the endoplasmic reticulum (17). It consists of three regions: a positively charged NH2-terminal region, a central hydrophobic core, and a polar COOH-terminal region (18). The amino acid coded by codon 10 of TGF-β1 gene is located in the central hydrophobic core and influences on the overall hydrophobicity of core sequence (4). Therefore, the change of a neutral proline to hydrophobic leucine residue disrupts the structure of the region, alters transportation across endoplasmic reticulum (4), and may affect the production or activity of TGF-β1.
The serum TGF-β1 levels were not measured in present study, because TGF-β1 is locally synthesized in the liver and may not reach systemic circulation for measuring. In addition, because platelet is the main source of serum TGF-β1, the serum levels of TGF-β1 do not always reflect its production in liver (5). Therefore, the analysis of TGF-β1 mRNA and protein expression in the liver are required for evaluating phenotypic differences according to the genetic polymorphisms.
TGF-β1 was also associated with the development of HCC (19) and the L/L genotype at codon 10 in TGF-β1 was associated with the development of HCC (7, 10). In the present study, HCC was present in 58% of patients of LC group, and the fact might influence on the results of this study. However, 70-90% of the patients with HBsAg positive HCC had already underlying cirrhosis (19). In addition, when cirrhotic patients without HCC were selected for analysis, the L/L genotype was still a significant risk factor for the development of cirrhosis.
Liver fibrosis is a highly complex disease process in which multiple genes interact with viral and environmental risk factors. Among them, age is a major risk factor for the development of cirrhosis irrespective of any etiology. In order to exclude the influence of age factor, present study's inclusion criteria restricted the age of patients over 50 yr old. The reasons why present study chose 50 yr old for age were; first, cirrhosis was generally diagnosed around this age (20) and second, it is not common that chronic HBV carriers remain chronic hepatitis without cirrhosis over this age in Korean population (personal experience). The longer duration of HBV infection is also an important risk factor of cirrhosis. Because most Korean patients with chronic HBV carriers were infected during perinatal period or early childhood, we could assume that the duration of HBV infection is nearly same as the patient's age. Present study showed no difference in the mean age of patients and in the duration of HBV infection between CH and LC groups.
Although initial HBV DNA levels were not known and the levels of HBV DNA were not serially followed up, the detectable HBV DNA was also a significant risk factor for cirrhosis in chronic HBV carriers. The assay for HBV DNA in the present study was the hybrid capture method, which can detect the level of HBV DNA over than 105 copies/mL. Comparing with real-time PCR method (lower detection limit: 25-50 copies/mL) for detecting HBV DNA, hybrid capture method is not sensitive enough to detect low level of viremia less than 105 copies/mL. However, hybrid capture method can detect the level of viremia that is considered as the threshold for active disease state and needed to be treated by current hepatitis B treatment guidelines (21).
The rate of cirrhosis development was higher not only in patients with continuous HBeAg-positive status compared to those with HBeAg seroconversion but also in patients with HBeAg reversion compared to those with sustained HBeAg soroconversion (22). These results indicated that the HBeAg was a strong risk factor for cirrhosis development. On the contrary of these reports, the annual incidence of cirrhosis was higher in HBeAg-negative patients than in HBeAg-positive patients (22). In Asian patients, cirrhosis and liver complications developed many years after HBeAg seroconversion (23). These contradictory results were addressed by taking into account both HBeAg status and HBV DNA level simultaneously in the evaluation of risk of cirrhosis development. HBeAg seroconversion without decreasing HBV DNA level was not a favorable marker for reducing fibrosis progression in chronic HBV carriers (24). Therefore, the significance of HBeAg as a risk factor for cirrhosis must be explained with HBV DNA level. Furthermore, serum HBV DNA level over 104 copies/mL was associated with a significant risk for progression to cirrhosis independent of HBeAg status (25). Although the present study did not analyze the risk of cirrhosis according to detail HBV DNA level, the level of HBV DNA over 105 copies/mL rather than the HBeAg status was more important in the development of cirrhosis.
The present study had a limitation that chronic hepatitis without cirrhosis was not diagnosed pathologically. We tried to exclude cirrhosis by several clinical diagnostic tools. Chronic hepatitis without cirrhosis was defined as no signs of the radiologic and endoscopic evidence of cirrhosis and portal hypertension. In addition to these criteria, patients with platelet count less than 150,000/mL were excluded for ruling out subclinical cirrhosis, because 1) low normal limit of platelet count is 150,000/mL, 2) platelet count well reflects portal hypertension (26) and 3) the degree of fibrosis is associated with platelet count in patients with chronic hepatitis B (27). Almost all patients in CH group had aspartate aminotransferase to platelet ratio index (APRI) less than 1.0 except three patients (data not shown). Those three patients had APRI over 1.0 and were all P/L genotype. Therefore they did not influence the result of present study when proportions of P/P and L/L genotypes were compared between CH and LC groups. Although APRI is not a sensitive marker to diagnose cirrhosis, its value of less than 1.0 is excellent for excluding cirrhosis (28). However, there was still a possibility that some patients with subclinical cirrhosis were included in CH group. The degree of cirrhosis in those patients might be mild compared to patients in LC group.
Antiviral agents over 1 yr might influence to the progression of fibrosis or cirrhosis in chronic HBV carriers. Several reports showed that antiviral agents prevented fibrosis progression pathologically in chronic HBV carriers (29) and improved Child-Pugh score in patients with cirrhosis (30). Therefore, present study did not assign the chronic HBV carriers who were treated with antiviral agents over 1 yr to CH group.
In conclusion, the L/L genotype at codon 10 in TGF-β1 and detectable HBV DNA level over 105 copies/mL are risk factors for development of liver cirrhosis. The genotyping at codon 10 in TGF-β1 may be a useful screening tool for the detection of the risk of cirrhosis development in chronic HBV carriers.



 XML Download
XML Download