

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cisplatin is a major anti-neoplastic drug for the treatment of solid tumors, but dose-dependent renal toxicity limits the use of optimal doses of this drug. Cisplatin has intracellular toxic effects on proximal straight and distal convoluted tubules. And DNA damage, oxidative stress, apoptosis, and inflammation have been implicated in the pathogenesis of cisplatin-induced renal injury (1).
Recent studies have shown increased tissue content of inflammatory mediators together with inflammatory cell infiltration, suggesting that inflammation plays an important role in cisplatin-induced renal injury (2, 3). Although the precise inflammatory mechanisms are unknown, marked attenuation of cisplantin-induced renal damage by inhibition of TNF-α indicates that TNF-α has a central role in mediating cisplatin-induced inflammatory renal injury (4).
Apoptosis is generally linked to the stepwise activation of a set of proteolytic enzymes called caspases. Several mechanisms that activate caspases 8 and 9 or executioner caspase 3 are all known to be involved in cisplatin-induced tubular cell apoptosis and caspase inhibition markedly reduces kidney injury (5, 6).
Peroxisome proliferator-activated receptors (PPARs) are members of the nuclear hormone receptor superfamily of ligand-dependent transcription factors known to be critical in regulating diverse biological processes, such as lipid metabolism, adipogenesis, insulin sensitivity, immune response, cell growth, and differentiation (7-10). Recently, the renoprotective actions of ligands for PPARγ, such as thiazolidinediones (TZD), have been reported. Although activation of a PPARγ in inducing renal tubular cell apoptosis has also been suggested, Lee et al. (11) reported that the PPARγ agonist, rosiglitazone, has renoprotective activity by reducing NF-κB phosphorylation and TNF-α production in cisplatin-induced renal damage. Recent observations have suggested that in addition to inhibiting inflammatory cytokine production, IL-10 is also important in mediating a tissue protective effect of PPARγ activation in several other injury models (12, 13). In addition, IL-10 has been known to inhibit inflammatory and cytotoxic pathways in ischemic or cisplatin-induced nephrotoxicity (14). However, whether the protective effect of rosiglitazone, a PPARγ agonist, in ciplatin nephrotoxicity is IL-10-dependent has not been clearly demonstrated. In this study, we investigated the effect of a PPARγ agonist in a cisplatin-induced AKI mouse model, with a focus on inflammation and apoptosis.
MATERIALS AND METHODS
Animals and drugs
Male BALB/c mice (20-25 g), purchased from Orient (Charles River Korea, Seoul, Korea), had free access to water and chow before experiment. Animal care followed the criteria of the Animal Care Committee of Korea University for the use of laboratory animals in research. Cisplatin (Sigma-Aldrich, St. Louis, MO, USA) was diluted in normal saline to a final concentration of 2 mg/mL. And mice were given a single intraperitoneal injection of cisplatin (20 mg/kg), while control animals received the same volume of normal saline. To investigate the renoprotective effect of rosiglitazone, another group of mice were pretreated with rosiglitazone (10 mg/kg; Alexis Biochemicals, Lausen, Switzerland) or the same volume of vehicle (0.1% dimethyl sulfoxide [DMSO] in normal saline) through daily intraperitoneal injections for 3 days before cisplatin injection. Mice were sacrificed at 4, 24, 48, and 72 hr after cisplatin administration. Blood was collected by intracardiac puncture and both kidneys were processed for histologic examination, RNA and protein isolation.
Biochemical analysis and histologic examination
Blood urea nitrogen (BUN) and creatinine levels were measured using a Hitachi 747 automatic analyzer. Renal tissue samples were fixed in 4% buffered paraformaldehyde and embedded in paraffin. After deparraffinization, 5 µm sections were processed and stained with periodic acid-Schiff (PAS). Tubular damage was estimated by examining 8-10 high power fields (HPF; ×200) per section by using a scoring system based on the percentage of damaged tubules per field (1, <25%; 2, 25% to 50%; 3, 50% to 75%; and 4, >75%). The mean score of each animal was compared. For the immunohistochemical detection of monocytes and macrophages, we stained formalin-fixed and paraffin-embedded kidney sections with monoclonal antibody against F4/80 (Serotec, Oxford, UK). After deparaffinization, endogeneous peroxidase activity was blocked with 0.5% hydrogen peroxidase. Sections were blocked for 10 min with goat serum, followed by incubation with primary antibody for 1 hr at room temperature (1:100). Specific labeling was detected with a biotin-conjugated goat anti-rat immunoglobulin G and an avidin-biotin peroxidase complex (Vector Laboratories, Burlingame, CA, USA). Naphthol AS-D chloroacetate (Sigma-Aldrich) was used to identify neutrophils. Eight to 10 HPFs were captured and the mean number of esterase-positive cells in the field was calculated in order for quantification.
Detection of apoptosis
Detection of apoptotic cells in the kidney was performed in paraffin-embedded kidney tissue sections using an Apop-TagPlus (Intergen, Purchase, NY, USA) following the manufacturer's protocol. The number of apoptotic cells in the outer medulla was semiquantitatively measured by counting 8-10 HPF (×200) per section.
Quantification of cytokines and chemokine by cytometric bead array
Quantification of various cytokines and chemokine in kidney tissues was done using cytometric bead array. A mouse inflammation kit (BD Biosciences, San Diego, CA, USA) was used according to the manufacturer's instructions to simultaneously detect mouse IL-12p70, TNF-α, IFN-γ, MCP-1, IL-10, and IL-6, as previously described (15). Briefly, a mixture of 6 capture bead populations (50 µL) with distinct fluorescence intensities (detected in FL3) coated with antibodies specific for the above cytokines and chemokine was mixed with each sample from kidney homogenates/standard (50 µL). Standard dilutions and test samples were added to the appropriate sample tubes (50 µL/tube). Additionally, PE-conjugated detection antibodies (detected in FL-2; 50 µL) were added to form sandwich complexes. After 2 hr of incubation at room temperature, the samples were washed with 1 mL of wash buffer and centrifuged (200 g for 5 min). Three hundred µL of wash buffer was added to each assay tube before acquisition in a FACScan flow cytometer (FACSCalibur™; BD Biosciences) and the sample results were analyzed using CBA software (BD Biosciences). The concentration for each cytokine in cell supernatants was determined by interpolation from the corresponding standard curve and normalized according to protein concentrations. The range of detection was 20-5,000 pg/mL for each cytokine.
Measurement of caspase activity
The activities of caspases 3, 8, and 9 in kidney tissues were determined by fluorometric detection of free 7-amino-4-trifluoromethylcoumarin (AFC; caspases 3 and 8) or 7-amino-4-methylcoumarin (AMC; caspase 9), following the manufacturer's protocol (ApoAlert™. Caspase Fluorescent Assay Kits; BD Biosciences, Palo Alto, CA, USA) by using a Synergy™ HT Multi-Detection Microplate Reader (Biotek, Woburn, MA, USA). Briefly, kidney tissues were homogenized in 1 mL of lysis buffer, incubated on ice for 10 min and centrifuged at 15,000 g for 10 min at 4℃. Supernatant containing 500 µg of protein was incubated for 1 hr at 37℃ in the presence of reaction buffer, 1 mM/L dithiothreitol (DTT), and 50 µM/L AMC or AFC substrate conjugates. The fluorescence was read at 400/505 (excitation/emission) nm for caspases 3 and 8 and 380/460 nm for caspase 9 and the samples were run in triplicate. The activity of the caspases was expressed as the percent increase compared to the saline-treated control group and samples which had caspase inhibitors served as negative controls.
RESULTS
Effect of rosiglitazone on kidney injury following cisplatin administration
Renal function was assessed at 4, 24, and 72 hr and histologic kidney damage was compared at 72 hr. As shown in Fig. 1, cisplatin injection significantly increased BUN and creatinine levels. In contrast, rosiglitazone pretreatment significantly reduced the increase of BUN and creatinine; histologic damage was less severe at 72 hr (Figs. 1, 2). Cisplatin caused renal damage with loss of the brush border, necrosis of tubular cells, cast formation. Rosiglitazone pretreatment significantly reduced these changes with improvement of the tubular injury score (Fig. 2).
Effect of rosiglitazone on cytokine and chemokine expression and inflammation
We investigated the effect of rosiglitazone on kidney cytokine and chemokine protein expression by CBA. Proinflammatory cytokines (TNF-α, IL-6, and IFN-γ) and chemokine (MCP-1) all increased significantly in cisplatin-treated animals at 24 hr. Rosiglitazone pretreatment attenuated these changes. In contrast to these proinflammatory mediators, IL-10, a potent anti-inflamamtory cytokine was significantly decreased in cisplatin-treated kidneys compared to sham. In rosiglitazone-pretreated kidneys, the kidney IL-10 protein level increased significantly. At 24 hr after cisplatin administration, renal function and histologic damage of the kidney was not apparent. Increased IL-10 production by rosiglitazone pretreatment was considered responsible for subsequent protection at 72 hr (Fig. 3). By immunohistochemical staining of kidney macrophages and neutrophils, kidney macrophages (F4/80 positive cells) and neutrophils (esterase-positive cells) increased markedly in cisplatin-treated kidneys; and rosiglitazone pretreatment also significantly reduced infiltration of these cells (Figs. 4, 5).
Effect of rosiglitazone on caspase activation and apoptosis
The effect of rosiglitazone on activation of different types of caspases and resulting apoptosis was examined. Activities of caspases 8, 9, and 3 in kidney tissues were measured and tubular cell apoptosis was assessed. The activity of caspase 3 increased significantly and rosiglitazone pretreatment attenuated the activation of caspase 3 at 24 and 72 hr. However, caspase 8 and 9 showed a low degree of activation following cisplatin administration, and rosiglitazone pretreatment did not have any effect on their activities (Fig. 6). This result suggested that caspase 3 were activated independent from caspase 8 or 9 by cisplatin and that caspase 3 was directly inhibited by rosiglitazone. We also performed TUNEL staining for quantifying renal tubular cell apoptosis and observed that increased tubular cell apoptosis by cisplatin were significantly attenuated by rosiglitazone pretreatment at 48 and 72 hr (Fig. 7).
DISCUSSION
In the present study, we demonstrated that pretreatment with the PPARγ agonist, rosiglitazone, attenuated cisplatin-induced kidney injury by reducing tissue inflammation and apoptosis. More importantly, rosiglitazone pretreatment increased the kidney IL-10 level at 24 hr, and this change was accompanied by decreased inflammation and cytotoxicity at 72 hr after cisplatin administration. Rosiglitazone also decreased cisplatin-induced tubular cell apoptosis by directly inhibiting caspase 3 activation.
Cisplatin, a major anti-neoplastic drug of solid tumors, has multiple intracellular effects, such as direct toxicity with reactive oxygen species, by activating mitogen activated protein kinases (MAPKinase), by inducing apoptosis, and by stimulating inflammation and fibrogenesis (1). Many potential therapeutic approaches targeting these steps for the prevention of cisplatin-induced renal injury has been extensively studied. In particular, recent evidence indicates that inflammation has an important role in the pathogenesis of cisplatin-induced renal injury. Ramesh et al. (4) demonstrated that TNF-α played a central role in the activation of various cytokines. They showed the beneficial effect of high dose salicylate was mediated by its anti-inflammatory effect in attenuating TNF-α production through reducing NF-κB transcription (16). The role of inflammation in cisplatin nephrotoxicity has also been confirmed by showing the protective effect of exogenous administered anti-inflammatory cytokine IL-10 (14). In our study, we also showed cisplatin evoked substantial tissue inflammation with upregulation of various inflammatory mediators. Unlike other studies that demonstrated transcriptional upregulation of inflammatory mediators, we measured tissue protein levels of various cytokines and chemokines early after cisplatin administration and observed that in addition to TNF-α, other cytokines, such as IFN-γ and IL-6, and the chemokine, MCP-1, are markedly increased in cisplatin-treated kidneys. In contrast to proinflammatory mediators, however, the IL-10 protein level decreased markedly in cisplatin-treated kidneys. These findings suggested that decreased endogenous IL-10 release might contribute to enhanced inflammation and subsequent tissue injury by cisplatin, because IL-10 is a well-known anti-inflammatory cytokine that has been known to suppress proinflammatory cytokines and chemokines. Rosiglitazone pretreatment, however, in contrast to cisplatin-treated kidneys, substantially improved endogenous IL-10 release with simultaneous suppression of TNF-α, IL-6, MCP-1, and IFN-γ. Rosiglitazone is a PPARγ agonist that has been reported to have an anti-inflammatory action in addition to its modulatory effect on glucose and fatty acid metabolism. A large body of evidence demonstrated that inhibition of transcription factors, such as NF-κB, NF-AT, Sp1, and AP-1 by a PPARγ is directly responsible for attenuating proinflammatory cytokine production (17-19). However, our results support that upregulation of IL-10 may also be responsible for the anti-inflammatory effects of a PPARγ. Although we did not provide a direct causal link between IL-10 and other proinflammatory mediators in this study, IL-10 might be critical in the protective mechanism, given that IL-10 has PPARγ-activated receptor response element (PPRE) promoter-specific for PPARs, and that rosiglitazone has been shown to increase IL-10 production from mature dendritic cells or CD4+ T cells (15). Additionally, we also observed the IFN-γ level increased markedly early after cisplatin injection. Interferon-γ is a well-known T cell cytokine that is known to play an important role in tissue injury. Liu et al. (20) demonstrated the pathophysiologic role of T lymphocytes by observing loss of protection in cisplatin nephrotoxicity in nu/nu mice by adoptive transfer of T lymphocytes. Although they identified infiltration of CD3+ cells early after cisplatin administration, they did not detect an increased IFN-γ level. Our study showed that IFN-γ increased markedly early in cisplatin-treated kidneys and that rosiglitazone pretreatment significantly suppressed IFN-γ production. Discrepancy in tissue cytokines and chemokines between experiments might come from using animals of different strains, different doses of cisplatin, or sample timing. In the experiments of Liu et al. (20), they observed that CD3+ cell infiltration peaked at 12 hr, returned to a normal level at 24 hr, but they measured tissue cytokine levels at 24 and 72 hr; and this timing might be too late for the detection of cytokine production. Although we did not demonstrate the morphologic evidence of T cell infiltration, increased production of IFN-γ from T cells is likely to contribute to the pathogenesis of cisplatin nephrotoxcity. The cisplatin-induced tissue cytokine milieu that favors inflammation might have ultimately led to substantial inflammation. We observed an increased number of F4/80-positive kidney macrophages and esterase-positive neutrophils 48 hr after cisplatin injection and also that rosiglitazone pretreatment markedly attenuated tissue inflammation. Neutrophils are thought to contribute to tissue injury by releasing a variety of mediators. And several anti-inflammatory strategies, which result in a decrease in neutrophil infiltration, attenuated cisplatin-induced kidney injury. The number of F4/80+ cells increased markedly by cisplatin; and also rosiglitazone pretreatment significantly reduced their infiltration. However, the role of F4/80 kidney macrophages in cisplatin nephrtoxicity is still unclear. Recent study by Lu et al. (3) demonstrated that liposome clodronate injection for the depletion of macrophages did not attenuate cisplatin-induced kidney injury.
Apoptosis is now recognized as an important mechanism of cisplatin-induced renal injury. There are two major pathways that lead to apoptosis, both of which culminate in a common death program. Signals from the mitochondrial and death receptor pathways activate "initiator" caspases 9 or 8, respectively, and this activation subsequently cleaves and activates "executioner" caspase 3 (5, 21, 22). Tsuruya et al. (22) demonstrated direct involvement of the receptor-mediated pathway and Park et al. (21) has showed that cisplatin induces apoptosis via activation of the mitochondrial pathway. However, the present study demonstrated that cisplatin-induced activation of caspase 3 is mostly independent of caspase 8 or 9 activation and contribute to tubular cell apoptosis. Although the upstream signal that led to direct activation of caspase 3 is not clear in this study, a variety of factors, such as direct DNA damage, inflammatory mediators, or reactive oxygen species are all likely to contribute to activation of caspase 3 and apoptotic cell death. Rosiglitazone pretreatment significantly reduced the activation of caspase 3 and apoptosis without an effect on caspases 8 or 9, suggesting the independent effect of rosiglitazone on caspase 3. Additionally, because inflammatory mediators also can induce tubular cell apoptosis, rosiglitazone-induced IL-10 upregulation might also contribute to decreased apoptosis.
In conclusion, our study demonstrates rosiglitazone pretreatment protects mice from cisplatin nephrotoxicity. The beneficial effect is thought to be mediated by upregulation of endogenous IL-10 production by rosiglitazone with subsequent reduction in tissue inflammation and cytotoxicity. Rosiglitazone is currently clinically available for the treatment of insulin resistance and its anti-inflammatory effect can be used to prevent cisplatin nephrotoxicity in the treatment of solid tumors.
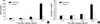






 XML Download
XML Download