

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Among the neonatal seizure syndromes, benign familial neonatal convulsions (BFNC), a rare autosomal dominant epilepsy characterized by unprovoked, generalized convulsions occurring in the first days of life, must be differentiated from non-epileptic paroxysmal events of the newborn and from other etiologies of neonatal seizures. The seizures are characterized by generalized hypertonia with apnea and cyanosis, followed by head movement and/or clonic or tonicclonic limb jerks (1). Most patients are seizure-free by 6 months of age, and psychomotor development in affected children is usually normal (2). Although BFNC represent a benign epilepsy syndrome, between 10% and 16% of patients experience seizure recurrence at a later time (1).
The idiopathic epilepsies, among which BFNC was listed, have been regarded as genetic in origin. This presumption was confirmed by the identification of mutations in KCNQ2 and KCNQ3 in families with BFNC.
We report here the first Korean family with BFNC. Five individuals in 3 generations were affected, and molecular analysis disclosed a KCNQ2 mutation.
CASE REPORT
Index case (IIId)
A female infant weighing 2.8 kg was born at 39 weeks, 2 days gestation to a 28-yr-old mother after an uncomplicated pregnancy. On the 3rd day of life, she presented with three brief generalized tonic seizures with upward deviation of the eyes and cyanosis of the lips which lasted approximately one minute. General and neurologic examinations were unremarkable. Electrolytes, calcium, glucose, cerebrospinal fluid examination, brain magnetic resonance imaging and electroencephalography (EEG) were all normal. She was discharged after 3 days without further seizures on phenobarbital. In follow-up, her phenobarbital was weaned at 3 months of age, but her seizures recurred 1 month later. Her EEG showed several spike discharges originating from the left and right central-temporal areas at that time. Phenobarbital was restarted and tapered off successfully without any seizure recurrence at 7 months of age. The child has had a normal neurodevelopmental course thereafter.
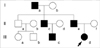
Detailed review of her family history revealed five affected family members, in three separate generations, who experienced similar neonatal convulsions, including the patient's grandfather, father, paternal uncle, and cousin (Fig. 1).
Patients
Ia (Grandfather): Patient Ia had 3 convulsions within the 1st month of life, but no further seizures after those initial episodes.
IIc (Paternal uncle): This patient experienced 5 convulsions within the first 3 months of his life. No further seizures were noted thereafter. The patient graduated from a postgraduate school.
IIe (Father): He experienced a single convulsion on the 2nd day of life. He also graduated from a postgraduate school and has no neurologic problems.
IIIc (A cousin): Eight months-old at the time of this writing. Although he experienced 6 convulsions within the 6 months of life, he has shown normal development.
In sum, despite multiple infantile seizures, no epilepsy or psychomotor dysfunction occurred in any affected family members even without the use of antiepileptic drugs.
To search for mutations in KCNQ2 in the pedigree, 16 exons and adjacent short intronic sequences of the gene were amplified by polymerase chain reaction (PCR) with DNA obtained from the affected individuals. Sixteen exons of the KCNQ2 gene and their intronic flanking sequences were amplified by PCR using a total of 19 sets of primers with exon 1 split into two and exon 6 into three primer pairs.
Genetic study revealed an R448X mutation located in the C-terminal segment of KCNQ2 in the family members with neonatal seizures (Ia, IIe, IIId, Fig. 2).
DISCUSSION
BFNC remains a diagnosis of exclusion, since many neonates present with cryptogenic convulsions associated with uncertain outcomes. Symptomatic neonatal seizures, secondary to hypoxic-ischemic-encephalopathy, metabolic disturbances or to an infectious process should be ruled out. Subsequently, the finding of self-limited neonatal seizures in multiple affected family members can clinch the diagnosis.
BFNC may be caused by mutations in KCNQ2 or KCNQ3. The KCNQ2 and KCNQ3 genes encode the heteromeric assembly of subunits through which muscarinic-regulated potassium current (M-current), a primary regulator of neuronal excitability, is established (3). The functional analysis of mutations thus far has indicated that a partial loss of activity of the K+ current is sufficient to produce epilepsy, and dominant negative mutations in KCNQ2 or KCNQ3 can result in a more severe phenotype (4).
In our case, the family history given by the grandmother of the proband was key to the diagnosis, and further genetic study identified a putative causative mutation in KCNQ2, the R448X (5, 6). Consistent with prior cases of R448X (5), none of our patients have experienced seizures outside of infancy. Interestingly, the KCNQ2 C-terminal truncation mutant R448X has been shown to reduce the potassium current by <50% and to cause no changes in the ion channel's biophysical properties (6). Given recent findings that ion channels may regulate diverse cellular functions in addition to their current conduction properties, this finding may suggest that another property of the KCNQ2 may be involved in its epileptogenic properties.
BFNC is an age-dependent, genetically determined epilepsy that typically resolves spontaneously. Kanaumi et al. (7) have demonstrated simultaneous high expression of KCNQ2 and KCNQ3 in human brain from late fetal life through infancy, coinciding with the time that BFNC occurs. Such developmental changes in potassium channel activity in the developing brain may explain the age-dependent expression of BFNC. However, inter-familial differences in susceptibility to subsequent febrile or afebrile seizures (1, 8) have been observed, and further genotype-phenotype correlations may facilitate our understanding of this condition.
Due to the immaturity of neonatal brain structures, analyzing the ictal phenomenology and EEG and definitive classification of BFNC is still difficult. On the other hand, BFNC shares features with benign rolandic epilepsy, the most common idiopathic localization-related epilepsy, including its age-dependency and presence of centrotemporal spikes (9, 10). In our study, the index case showed intermittent right or left centrotemporal sharp wave discharges, and the seizures observed were generalized tonic seizures. It is thought that as data accumulates, the electroclinical features of the ictal, interictal and follow-up EEG may yield insight into the mechanism of seizure generation.
In conclusion, we report the first Korean case of BFNC with genetic confirmation. The mutation was the same KCNQ2 as has been reported in other ethnic groups.

 XML Download
XML Download