

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acrodermatitis enteropathica (AE) is a rare autosomal recessive disease, which causes the classic clinical triad of acral dermatitis, diarrhea and alopecia (1). AE results from a defect in zinc absorption (1, 2), and clinical presentations can be resolved by oral zinc replacement. Recently, AE has been found to be caused by a mutation of the SLC39A4 gene, which encodes the ZIP4 zinc transporter and is located on chromosome 8q24.3 (3-5).
We encountered an 8-month-old boy with acrodermatitis enteropathica who improved with oral zinc supplementation. He was confirmed to have congenital zinc deficiency by detection of a mutation of SLC39A4. We report the first case of AE proven by detection of a mutation of SLC39A4 in Korea.
CASE REPORT
Clinical and biochemical analysis
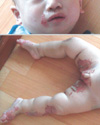
An 8-month-old boy visited the dermatologic outpatient clinic on 6 May 2008 because of skin lesions on his inguinal area and knees since 7 months of age. His body weight and height were 8.4 kg (25-50 percentile) and 69.2 cm (10-25 percentile), respectively. He had shown no evidence of delay in his growth. He had defecated a small amount of loose stool for 3 or 4 times per day one month ago. His diarrhea was wax and wane and the total amount of stool was less than 10 g/kg/day. He had been breast fed up to 2 months of age, and he was being weaned at the time of visit. After that first visit, the patient was treated with ointment and oral medication for atopic dermatitis. Despite treatment, however, the skin lesions progressed to his cheeks, feet and scrotum, and were accompanied by oozing. With a presumptive diagnosis of AE, he was treated with oral zinc replacement (zinc gluconate) for 11 days. During zinc replacement, the skin lesions improved. At that point, we stopped the zinc replacement for 3 weeks and considered acquired zinc deficiency. However, the skin lesions reappeared and progressed to erosion and eczematous changes over his hands, knees, feet and perioral and inguinal areas (Fig. 1). His plasma zinc level was 18.8 µg/dL (normal 66-110 µg/dL). The zinc replacement was restarted (3 mg/kg/day), the skin lesions improved after 1 week, and his plasma zinc level increased to 90.1 µg/dL. His hair growth started one month later. We suspected congenital zinc deficiency because of the recurrence of AE after the cessation of zinc supplementation. His parents are not consanguineous and have had only one baby, this patient. There is no family history of zinc deficiency.
Genetic analysis
To confirm the diagnosis of congenital zinc deficiency, a molecular genetic study on SLC39A4 was performed by direct sequencing analyses. We collected blood specimens from the proband and his family. Genomic DNA was isolated from leukocytes using the Wizard genomic DNA purification kit according to the manufacturer's instructions (Promega, Madison, WI, USA). All coding exons and flanking intronic sequences of SLC39A4 were amplified by polymerase chain reaction (PCR) using the primers designed by the authors (available upon request) on the Thermal Cycler 9700 (Applied Biosystems, Foster City, CA, USA). Direct sequencing was performed using the same primers for PCR on the BigDye Terminator Cycle Sequencing Ready Reaction kit (Applied Biosystems) on the ABI Prism 3130 genetic analyzer (Applied Biosystems). The sequence chromatograms obtained were compared to the reference sequences (NP_570901.2 and NM_130849.2) using the Sequencher software (Gene Codes Corporation, Ann Arbor, MI, USA).
Clinical and genetic findings
We found that the patient was compound heterozygous for 2 mutations of SLC39A4 (Fig. 2). One was a missense mutation in exon 2, leading to a substitution of the 95th amino acid residue arginine with cysteine (c.283C×T [p.Arg95Cys]). The other was a splice site mutation occurring in the consensus donor site of intron 7 (c.1287+2T×C). Family study revealed that his father and mother were heterozygous carriers of each mutation.
Currently, the patient takes oral zinc replacement (2 mg/kg/day) and shows normal skin integrity. His plasma zinc and copper levels are in the normal range. He shows normal growth and development.
DISCUSSION
Acrodermatitis enteropathica (AE) is an inherited, rare autosomal recessive disease, with an estimated incidence of 1 per 500,000 children without predilection for race or sex (6). The high rate of consanguineous marriages in some cohorts leads to a higher carrier state of the mutation (1:50) (7).
Clinical manifestations of AE include characteristic skin lesions, such as acral and periorificial erythematous, scaly plaques with crusts and ulcerations, diarrhea, alopecia, growth failure and delayed puberty (1, 2). AE-like skin eruptions can be developed by dietary zinc deficiency; these can be resolved by oral zinc replacement and do not recur (8). However, AE usually recurs after ceasing the supply of zinc (2).
At first, this patient was misdiagnosed with atopic dermatitis because of eczematous changes of his inguinal area and knees. After applying steroids, the lesions did not improve and had even progressed. We reassessed him as having dietary zinc deficiency and administered oral zinc gluconate. After 10 days, the skin lesions disappeared, and he had a good appetite and was well nourished. As a result, we stopped zinc replacement. However, his skin lesions relapsed and became aggravated within one month after cessation of the zinc. We then considered congenital zinc deficiency and checked for the mutation of SLC39A4.
Acrodermatitis enteropathica (AE) was first named in 1943 (9), and its pathogenesis was discovered in 1973 (2). In 2001, Wang et al. found the location of the AE gene on chromosome 8q24.3 by homozygosity mapping (3). In 2002, the defected gene was identified as SLC39A4 (for solute carrier family 39, member 4), which was known to encode the zinc/iron-regulated transporter-like protein (ZIP4) zinc transporter that included zinc-uptake proteins (4, 5). This gene is expressed in parts of the intestine, including the duodenum and jejunum, which are essential sites of zinc absorption. The protein product of SLC39A4 is dynamically regulated by zinc (4).
Molecular genetic studies revealed that our patient was heterozygous for 2 mutations in the SLC39A4 gene. SLC39A4 is composed of 12 exons, encoding a protein of 647 amino acids (MIM#607059). The Human Genome Mutation Database shows 25 different mutations in SLC39A4, including 15 missense mutations and 2 splicing mutations (Ref: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=SLC39A4). A recent report described other novel mutations and unclassifiable variations (10). Since most SLC39A4 mutations are private and occur in different types and over the entire gene, direct sequencing of all coding exons and flanking intronic sequences is the molecular diagnostic approach of choice in AE. The missense mutation in our patient, Arg95Cys, was previously reported by Nakano et al. (11) in a Japanese family with AE. Based on the observation that Arg95Cys was not occurring in 100 control chromosomes screened, they concluded that Arg95Cys was a disease-causing mutation. The precise functional implication of the Arg95 residue, however, needs to be determined (11). The other mutation, c.1287+2T>C, was a novel splicing mutation in the donor site of intron 7, following 2 mutations in acceptor sites of introns 2 and 6 found in the literature (11). Family study revealed that each of the mutations had been inherited from his parents, compatible with the autosomal recessive inheritance pattern of the disease. Partly due to the limited number of mutations reported thus far, no genotype-phenotype correlations have been reported in SLC39A4-related AE (10).
The treatment of AE is simple; only oral zinc replacement is needed (12). Zinc replacement therapy in AE should be started at 3 mg/kg/day of elemental zinc (12). Serum or plasma zinc levels and zinc-dependant enzyme levels should be monitored every 3 or 6 months (12). In addition, the copper level must be monitored, because zinc supplementation usually reduces copper absorption (12). The copper level in this case was within normal limits, 114.2 µg/dL (normal range, 75-145 µg/dL).
In summary, patients with relapsed nutritional AE after ceasing zinc supplemention should be tested for SLC39A4 gene mutations to confirm congenital zinc deficiency. We report the first case of congenital AE confirmed by molecular genetic study with a novel splicing mutation in the donor site of intron 7.

 XML Download
XML Download