

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cat-eye syndrome (CES) is a rare malformation syndrome characterized by a variable pattern of congenital anomalies. The characteristic features of CES include ocular coloboma, preauricular pits or tags, anal anomalies, and congenital heart and renal malformations. Furthermore, CES may be associated with other craniofacial malformations, skeletal anomalies, and mental retardation (1, 2). CES is generally associated with a supernumerary marker chromosome containing duplicated material of chromosome 22. The marker is normally bisatellited and isodicentric, which causes a tetrasomy of the p arm and part of 22q11.1 (1, 3).
The proximal portion of the long arm of chromosome 22 has been recognized as a hot spot for chromosomal rearrangements, and contains both the CES and DiGeorge syndrome critical regions (4, 5). Multiple congenital malformations evidencing overlap with CES can arise from der(22) syndrome (6) and the interstitial duplication of proximal 22q (7, 8).
Although one Korean case with partial trisomy 22q has been reported, this case belonged to imbalanced chromosomal anomaly caused by maternal balanced translocation rather than CES in the strict sense (9). There has been no report of typical CES cases confirmed by cytogenetic and molecular cytogenetic analyses in Korea yet. In this study, we present a Korean boy with CES resulting from a supernumerary marker chromosome, which was confirmed as a bisatellited and isodicentric chromosome 22 via cytogenetic and molecular cytogenetic analyses, including karyotyping, array comparative genomic hybridization (array-CGH), and fluorescence in situ hybridization (FISH).
CASE REPORT
A 2-month-old Korean boy was referred to the clinical genetics clinic of Ajou University Hospital for the evaluation of an abnormal karyotype at October 21th, 2009. The patient was born at the 37th week of gestation after an uneventful pregnancy. The parents were healthy and unrelated, and he was the first baby to both parents. At the patient's birth, the mother and father ages were 38 and 42 yr of age, respectively. The patient's birth weight was 2,570 g (10-50th percentile) and his birth length was 47.0 cm (10-50th percentile). On prenatal ultrasonography, a dilatation of the left ureter was observed, and hydronephrosis was suspected. After a single umbilical artery was identified and follow-up ultrasonography revealed a small dysplastic left kidney with a markedly dilated left ureter and ureterocele, the patient was admitted to our neonatal intensive care unit, and chromosome analysis was conducted on the 1st day after birth. Physical examination revealed characteristic facial features including low set ears, mild hypertelorism, down-slanting palpebral fissures, flattened nasal bridge and tip, micrognathia, and bilateral micropthalmia with iris colobomata. However, preauricular pit and ptosis were not detected. Echocardiography revealed a secundum atrial septal defect (ASD) with a small left-to-right shunt, and the result of the transient evoked otoacoustic emission test evidenced a reduced response on the left ear, and suggested the presence of effusion in the middle ear. Left undescended testis and micropenis with stretched length of 1.5 cm was also observed, however, no anal atresia was present.
A detailed opthalmological examination was conducted when the patient was 3.5 months of age, and bilateral microphthalmia and bilateral inferior colobomata of the iris, choroid, and retina were observed. In both eyes, the optic disc and macula were not visible due to chorioretinal colobomata. Mild lens opacity of the left eye and pendular-type nystagmus of both eyes was also noted. Brain MRI revealed bilateral multiple colobomata in both eyeballs, but no abnormalities in the bilateral optic nerves were noted. The visual evoked potential findings also suggested that bilateral ocular lesions were present.
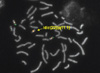
Chromosome analysis was conducted in accordance with standard methods on cultured cells from peripheral blood samples obtained from the patient and both parents. Metaphase preparations were analyzed via three different staining techniques (GTG, CTG, and Nucleolar Organizing Region). The results of chromosome analysis of 40 cells revealed the presence of a supernumerary bisatellited and isodicentric marker chromosome, which was apparently derived from an acrocentric chromosome (Fig. 1).
This marker chromosome was analyzed further via array-CGH with BAC clones. Commercially available array-CGH slides (MACArray Karyo 1440 BAC-chip, Macrogen, Seoul, Korea) were analyzed using a chromofluor image analysis system (Array scanner, Array analysis, Macrogen). The slides contained 1,440 human BAC clones including 356 cancer-related genes from BAC libraries at a resolution of 2.3 Mb. Each BAC clone was represented on an array as triplicate spots, and each array was scanned with a GenePix4000B scanner (Axon Instruments, Foster City, CA, USA) and subsequently analyzed using array software (MAC VIEWER, Macrogen). Green (test) to Red (reference) (G/R) ratios were determined automatically for each sample, and the normalized G/R ratio was considered to represent the relative average number of copies of the sequence for those spots that were selected as controls. Spots with G/R ratios greater than the mean plus 2.0 standard deviations (1.20) were regarded as amplifications or gains of the indicated copy number; spots with G/R ratios less than the mean minus 2.0 deviations (-0.80) were regarded as losses of the copy number. The result of array-CGH analysis in this patient showed duplication at 22q11.1 (Fig. 2). The size was estimated as approximately 0.4 Mb (genomic position 15,500,000-15,900,000).
In order to confirm this duplication, FISH analysis of this marker chromosome was conducted on interphase and metaphase spreads with a probe for the CES critical region (D22S43, Macrogen) on 22q11.1, and confirmed a duplication of the CES critical locus on 22q11.1 (Fig. 3). However, the FISH probe for the DiGeorge critical region (TUPLE1, Vysis, Downers Grove, IL, USA), localized to 22q11.2, exhibited no signal on the marker chromosome. These results indicated that this marker chromosome was derived from two different 22 chromosomes, with break points located on the q11.1 of both chromosomes.
Therefore, the karyotype of the patient was defined as 47,XY, +mar.ish idic(22)(q11.1)(D22S43+).arr 22q11.1(15,500,000-15,900,000)x4, resulting in tetrasomy of the p arm and the proximal part of 22q11. Cytogenetic analysis of both parents demonstrated a normal karyotype without numerical or structural abnormalities of chromosome 22, thereby indicating that the marker chromosome had arisen de novo.
DISCUSSION
Although CES is a rare malformation syndrome with an estimated incidence of between 1:50,000 and 1:150,000 (10), the clinical phenotypes of CES are quite distinct. The principal clinical features of CES include anorectal malformations, urogenital malformations, ocular colobomata, preauricular skin tags and/or pits, and congenital heart defects. Minor features include down-slanting palpebral fissures, hypertelorism, skeletal malformations, dysplastic ears, micrognathia, and microphthalmia. Neuropsychological development could be normal to severely retarded, and other neurological signs such as impaired ocular motility, hearing difficulty, spasticity, ataxia, and seizure are frequently identified in these cases (11). However, most of the features associated with CES are variably expressed even in familial cases (12). Only 41% of published CES patients show the classical triad of symptoms (2)-iris coloboma, anal anomalies, and preauricular malformation-and less than 10% of patients present all three major clinical features (11). In our case, ocular colobomata were revealed only among the classical triad, and anal and preauricular anomalies could not be detected. Although preauricular and anal malformations are the most frequently observed signs, and observed in 70-80% of other reports (2, 11), our case exhibited typical ocular findings including multiple colobomata, micropthalmia, and strabismus, as well as urogenital anomalies such as left kidney dysplasia, cryptorchidism, and micropenis, in addition to facial dysmorphism. These physical findings of our patient were sufficient for a clinical diagnosis of CES. Congenital heart defects are also commonly associated findings observed in 50-60% of CES patients (2, 11). In our case, a secundum ASD was detected via echocardiography, whereas total anomalous pulmonary venous return (TAPVR) and tetralogy of Fallot have been reported as more frequently presenting anomalies in cases of CES (11). The correlation between the clinical severity and size of the marker chromosome has yet to be clearly determined (4).
Classical CES is usually associated with a supernumerary bisatellited marker chromosome 22, which results in partial tetrasomy 22 (3). Three types of CES have been described, and these types are differentiated by the localization of the two recurrent CES breakpoints (13). Thus, type I CES chromosomes are symmetrical with two proximal breakpoints, but do not contain the DiGeorge critical region. Type II CES chromosomes harbor one (type IIa) or two (type IIb) distal breakpoints, which result in one or two additional copies of the DiGeorge critical region (5, 14). In our case, FISH studies showed that the marker chromosome harbored two copies of the CES critical region, but did not harbor the DiGeorge critical region. Therefore, our patient could be diagnosed with type I CES. No obvious difference has been determined to exist between the phenotypes of type I or type 2 CES patients, although relatively few patients have been studied thus far (14). This suggests that the duplication of the region deleted in cases of DiGeorge syndrome would induce a subtle phenotype.
Homologous recombination events between the breakpoints on 22q11 during meiosis have been implicated previously in CES rearrangements (15), and the U-type exchange of between sister chromatids and homologous chromosomes would induce the formation of the inv dup (22) of CES, as well as an eccentric fragment that would be lost (16). Fourteen genes have thus far been identified in the CES critical region located in the most proximal 2-2.5 Mb of 22q11. Two of these genes, CECR1 and CECR2, have been considered as candidates for involvement in the duplication phenotype (4). The CECR1 gene was expressed in the outflow tract and atrium of the heart and in the VII/VIII cranial nerve ganglion, suggesting its potential involvement in CES heart and facial defects (17). The other gene, CECR2, encodes for a putative transcriptional coactivator involved in neurulation and chromatin remodeling, particularly during later brain development and in the eye (18).
Here, we report a Korean CES patient, who showed partial tetrasomy of chromosome 22q11.1 resulting from a supernumerary isodicentric marker chromosome. Cytogenetic and molecular cytogenetic analyses led to the confirmative diagnosis by identifying the origin of the marker chromosome first in Korea.


 XML Download
XML Download