

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Familial juvenile hyperuricemic nephropathy (FJHN, OMIM #162000) is a rare autosomal dominant disorder characterized by hyperuricemia, gout and chronic kidney disease (1-4). It was first described by Duncan and Dixon in 1960 and more than 50 families in various ethnic groups have been described (5). However, there has been no report about precise incidence and prevalence of FJHN. The hyperuricemia, which is associated with decreased urinary excretion of urate, is known to causes chronic kidney disease in most patients. Affected family members show the impairment of urate excretion before puberty and usually develop hyperuricemia and gout after adolescence (6). Renal function gradually deteriorates and results in end-stage renal disease within 10 to 20 years. Diagnosis is suggested by a fractional excretion of uric acid of <5% (normal, 10-15%) with the symptoms and signs of FJHN (7).
The gene causing FJHN has been mapped to chromosome 16p11-p13 and in close proximity to the gene for medullary cystic kidney disease type 2 (MCKD2, OMIM #603860) (2). Mutations of the gene encoding uromodulin (UMOD) have been reported in several studies about various ethnic FJHN families. Eiji et al. reported five separate heterozygous missense mutations of the UMOD gene in five Japanese families with FJHN (1). Stacey et al. pursued linkage studies in seven European families with FJHN. But two of seven families were found not to be linked to chromosome 16p11-p13, thereby demonstrating genetic heterogeneity in more than 25% of FJHN families (8). Its clinical and histologic features are similar to those of MCKD type 2. Recent genetic studies revealed that MCKD type 2 is also associated with mutations in the UMOD gene, therefore an opinion is emerging that FJHN and MCKD type 2 are not the different diseases (9).
It is not clear what pathologic role of mutations of UMOD gene as a cause of renal urate under-excretion in FJHN. We found a Korean family with FJHN and five male members of them had gout and chronic kidney disease, which were diagnosed at teenage. There has been only one FJHN case report of Korean family without genetic analysis (10). This is the first report of genetically diagnosed FJHN in Korea.
CASE REPORT
A 16-yr-old male had been suffered from waxed and waned symptoms of swelling and pain of right 1st metatarsal joint since his age of 14. Recently he was hospitalized at a local clinic due to same symptoms, and was diagnosed as hyperuricemia, gouty arthritis and renal insufficiency. Though his symptoms were resolved with prednisolone, he was transferred to nephrology department of our hospital for further evaluation on abnormal renal function (April 4, 2009). On admission, he reported stable vital signs and no symptoms associated with azotemia. On physical examinations, he had no peripheral edema. Electrocardigraphy and simple radiography showed normal findings. The blood urea nitrogen was 17.5 mg/dL, serum creatinine was 1.59 mg/dL. Laboratory tests revealed hyperuricemia (uric acid 11 mg/dL) and renal under-excretion of urate (fractional excretion of uric acid 3.13%; 24 hr urine uric acid 327 mg). Serum total protein was 7.6 g/dL, albumin was 4.7 g/dL and total cholesterol was 161 mg/dL. Other laboratory tests showed results within normal range. There were no abnormal urinary findings like hematuria, proteinuria and pyuria. Abdominal sonography showed normal size and echogenicity in both kidneys, and no other structural abnormality was found. A renal biopsy was not performed.
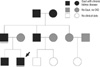
In the family history, his elderly brother was diagnosed with gout and renal insufficiency at age 18. Kidney biopsy, which was performed at his age of 20 in one hospital, revealed tubulointerstitial disease. His grandfather, father and uncle were also diagnosed with gout and chronic kidney disease at teenage and they have been treated with allopurinol for several years (Fig. 1). All of them had no other chronic disease such as hypertension or diabetes.
Two peripheral blood samples for gene analysis were obtained from patient and his father. DNA sequence analysis of the 10 exons of the UMOD gene was undertaken for genetical confirmation of FJHN. Gene analysis revealed a novel heterozygous missense mutation (c.1382C>A, p.Ala461Glu) that altered evolutionary conserved residues in the gene encoding UMOD (Fig. 2). Detected mutation was located in exon 6. The patient has been treated with allopurinol (100 mg/day) since admission and patient's serum uric acid level begun to decrease. Ten months later, his serum uric acid level was 6.4 mg/dL and serum creatinine level was 1.63 mg/dL. He remained clinically asymptomatic throughout this period. In this family, other affected members were treated with allopurinol and their serum uric acid levels were maintained. None of them developed end stage renal disease.
DISCUSSION
In most but not all families with FJHN, genetic studies have revealed mutations in the UMOD gene located on chromosome 16p11-p13. Many studies revealed various heterozygous missense mutations in the UMOD gene of families with FJHN. Uromodulin, which is also called Tamm-Horsfall protein, is the most abundant protein in normal urine and a major component of urinary casts. Uromodulin is synthesized exclusively and abundantly in the thick ascending limb of the loop of Henle and is known to have a pro-inflammatory potential such as activation of neutrophil, stimulation of monocyte to proliferate and release cytokines and gelatinases. UMOD gene knockout mice showed difficulty in clearing bacteria from the urinary bladder (11). Several studies have demonstrated reduced levels of UMOD in the urine of patients with FJHN and other studies have reported tubulointerstitial immune complex nephritis in rats immunized with UMOD protein (12, 13). Therefore, in the past it was simply thought that pro-inflammatory potential of UMOD protein might be related with the tubulointerstitial nephritis in FJHN. However, another recent study revealed that patients with FJHN who did not have UMOD mutations still showed reduced urinary excretion of uromodulin (14). Gersch et al. (15) also reported that UMOD knockout mice did not develop hyperuricemia. However, precise functions of UMOD still remain obscure and it is also not clear how mutations in the UMOD gene affect decreased urate excretion in patients with FJHN. Karin et al. reported there was a markedly increased expression of UMOD in a cluster of tubule profiles, suggesting an accumulation of the protein in tubular cells in families with FJHN and they also showed decreased urinary excretion of wild-type uromodulin (12). Jennings et al. (16) recently reported wild type and mutant uromodulin cDNA constructed in polarized monolayers of cultured kidney cell and found that both wild type and mutant uromodulin protetin were secreted more on the apical than the basolateral side of the monolayers . The author also reported the apical secretion of the mutant uromodulin was reduced, whereas the basal secretion was unaffected, suggesting that the mutant uromodulin in kidneys of patients with FJHN might elicit an immune response to uromodulin, which results in tubular injury and interstitial fibrosis.
Controversy exists as to whether lowering serum uric acid slows the progression of renal failure; the studies reporting benefit have usually involved starting a xanthine oxidase inhibitor early in the course of the disease (8). In the present FJHN family, a single nucleotide substitution was found to cause a heterozygous missense mutation (c.1382C>A, p.Ala461Glu) in exon 6, which had not been described previously. The present family members with gout and chronic kidney disease have been treated with allopurinol during several years and their serum uric acid levels have been maintained within normal range. None of them developed end stage renal disease. However, genetic analysis was not performed in unaffected members of the present FJHN family. In the present FJHN family, all five male members with hyperuricemia and chronic kidney disease were treated with allopurinol and their serum uric acid levels were maintained. All of them showed reduced renal function (40-60 mL/min/1.73 m2), however none of them developed end stage renal disease.
In conclusion, we observed a FJHN family confirmed by genetic analysis for the first time in Korea. Genetic analyses show UMOD gene mutation, a novel heterozygous missense mutation (c.1382C>A, p.Ala461Glu), which has not been reported previously.

 XML Download
XML Download