

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cystic fibrosis (CF) is caused by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR; ABCC7) gene located on the long arm of chromosome 7 (1). The gene product of CFTR forms an anion selective ion channel which is required for the normal function of epithelia lining the airways, intestinal tract, pancreatic duct, and vas deferens. Clinical features of CF include an exocrine pancreatic insufficiency and male infertility; however the major cause of morbidity and mortality is caused by pulmonary diseases (2).
In addition to the presence of typical clinical features, the diagnosis of CF is assisted by several other findings, such as a history of CF in a sibling, positive sweat Cl- test, identification of CFTR mutations, and an abnormal nasal potential difference (4). CF is the most common life-threatening genetic disease of Caucasians and its incidence in Europe is approximately 1 in 1,000-3,000 newborns (5). CF is rare in the Korean population. Thus far only a few cases have been reported (6-9). Here, we report a Korean CF patient who was diagnosed by both typical clinical features and a positive sweat chloride test. The genetic analysis of patient's CFTR gene and the related molecular functional study revealed that the patient had a pathogenic L441P mutation in one allele.
CASE REPORT
A 5-yr-old girl was admitted to Sanggye Paik Hospital due to chronic productive cough and greenish sputum. Eight months prior to the current hospitalization, she developed cough, sputum, rhinorrhea, and fever which were associated with frequent respiratory tract infection.
She was born by vaginal delivery. A meconium staining of the amniotic fluid was observed at delivery, but the evidence of meconium aspiration was not clear. Her birth weight was 2.45 kg, which was small for her gestational age (40 week). She had an older brother while the CF in family history or CF-related symptoms were found negative from her older brother. The patient had been suffered from diarrhea persisted from 3 months of age, which led to hospitalization and poor weight gain since then. She had been hospitalized for three times from 2 yr of age due to recurrent pneumonia and respiratory problems. Her body weight and height on admission were 12.2 kg (<3 percentile) and 95.9 cm (10-25 percentile), respectively.
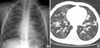
Chest radiographs and computed tomographic (CT) scans showed bilateral diffuse bronchial wall thickening and symmetric bronchiectasis in both lung fields (Fig. 1). The diseases caused by immunodeficiency or allergic reactions were ruled out by her normal complete blood cell count, serum immunoglobulin levels and specific IgE levels. P. aeruginosa was cultured from her sputum. Abdominal ultransound and pancreaticobiliary CT scan revealed moderate to severe fatty liver and atrophy of the pancreas. Because she had chronic diarrhea, poor weigh gain, and atrophy of the pancreas as well as pulmonary symptoms, the patient was suspected to have CF.
To confirm the diagnosis, the sweat chloride concentration was measured by a quantitative pilocarpine iontophoresis sweat test recommended by the National Committee for Clinical Laboratory Standards, USA (National Committee for Clinical Laboratory Standards. Sweat Testing: Sample Collection and Quantitative Analysis. Villnova, Pa: National Committee for Clinical Laboratory Standards; 1997 NCCLS document C34-A.). The test was repeatedly performed at Yongdong Severance Hospital, Yonsei University on two separate days. The average sweat chloride concentrations on both forearms were 78.3 mM/L and 99.0 mM/L in two separate measurements with a one-week interval. These values were over the reference limit (<40 mM/L) and highly suggestive of CF.
An initial genetic screening was performed on the 11 mutation/polymorphism loci of CFTR gene previously identified in the Korean population (10) and on the 10 most common disease-associated loci in Caucasians using the SNaP Shot method (Applied Biosystems, Foster City, CA, USA). However, none of the above mutations were identified in the patient's gene. Therefore, we next scanned the genetic variations in the CFTR gene using the denaturing gradient gel electrophoresis (DGGE) and subsequent nucleotide sequencings to find unknown CFTR mutations as previously detailed (10). Interestingly, a non-synonymous L441P mutation of CFTR was identified in one allele by DGGE and consecutive nucleotide sequencings (Fig. 2). Although we performed a comprehensive search for the entire coding regions and exonintron splicing junctions of CFTR gene, no additional mutations were found. Among family members, patient's mother was available for genetic tests. However, no CFTR mutations including L441P were found in the blood samples from patient's mother (Fig. 2). Therefore, the L441P mutation was assumed to come from patient's father.
It has been reported that the L441P mutation of CFTR is associated with CF in Japan and its allele frequency was estimated around 4.5% among CF patients in Japan (5). However, the molecular pathogenic mechanisms of the L441P mutation of CFTR are currently unknown. Therefore, we investigated the disease-causing mechanism of L441P using an integrated molecular and physiologic study. The CFTR plasmid carrying a L441P mutation was constructed by sitedirected mutagenesis using QuickChange kit (Stratagene, La Jolla, CA, USA) with pCMV-CFTR plasmid according to the manufacturer's protocol. The residue thymine base at 1454 of CFTR was substituted with cytosine and the mutagenic sequence was confirmed by the DNA sequencing. The total amount of CFTR protein was compared in HEK 293 cells transfected with plasmids for wild type CFTR or CFTR carrying the L441P mutation by immunoblotting of total cell lysates. In addition, the amount of surface-expressed proteins was examined by the immunoblotting of surface biotinylated proteins.
During secretory pathway, the polypeptidic chain of CFTR undergoes post-translational modifications at its glycan moiety in the Golgi complex to produce the fully glycosylated mature form, also known as band C, of about 170-180 kDa (11). In immunoblotting of cell lysates, most of the wild type CFTR protein was detected as the fully glycosylated mature form, whereas virtually all of L441P mutant proteins appeared as the ER core-glycosylated form of about 150 kDa, also known as band B (Fig. 3). These results imply that the L441P mutant CFTR protein has a defect in the ER-to-Golgi trafficking of the secretory pathway of membrane protein. Consequently, the surface biotinylation results revealed that the L441P mutant protein failed to reach the plasma membrane, whereas the fully glycosylated form of wild type CFTR was expressed on the cell surface (Fig. 3).
To investigate the intracellular localizations of the mutant CFTR, immunostaining was performed in HEK 293 cells transfected with plasmids for wild type CFTR or CFTR carrying the L441P mutation. The CFTR protein was stained with anti-CFTR 24-1 antibodies (R&D Systems, Minneapolis, MN, USA) and fluorescein isothiocyante-conjugated secondary antibodies. The ER-resident calnexin protein was stained with anti-calnexin antibodies (Abcam, Cambridge, MA, USA) and rhodamine-conjugated secondary antibodies, and then images were obtained with a Zeiss LSM510 confocal microscope. As shown in Fig. 4, wild type CFTR was mainly localized on the plasma membrane (arrow heads). On the other hand, L441P mutant protein was found in the ER, which was confirmed by co-localization with calnexin, an ER membrane protein (Fig. 4).
It is well known that CFTR protein has a cAMP-activated Cl- channel function (2). Thus, the cAMP-activated chloride channel activities of CFTR-expressing HEK 293 cells were measured in the whole cell configuration. Treatments with the adenylyl cyclase activator forskolin (FSK) produced a large inward current in NMDG-Cl solutions (Fig. 5). The I-V relationship measurements revealed the typical CFTR current of a linear I-V relationship when a ramp pulse from -120 mV to +120 mV was applied at peak current (Fig. 5B). The current density of wild type CFTR at a -30 mV holding potential was 37.48±6.20 pA/pF. However, the cAMP treatment (5 µM FSK) failed to activate the Cl- currents in cells transfected with L441P mutant CFTR (Fig. 5).
DISCUSSION
CF is caused by the loss-of-function mutations in the CFTR gene and inherited as an autosomal recessive trait. The classical form of CF is characterized by the progressive lung disease, pancreatic dysfunction, elevated sweat electrolytes, and male infertility (12). However, a wide variability in the clinical presentation is found among patients. For example, only about 20% of affected infants are born with intestinal obstruction and meconium ileus. Other patients are diagnosed with various modes of presentation from birth to adulthood and with considerable variability in the severity and rate of disease progression. Although progressive lung disease is the most common cause of mortality in CF, there is great variability in the age of onset and severity of lung disease in different age groups. Effects from other genes and environmental factors are suggested to make important contributions to disease progression, because pulmonary phenotype varies even within the same CFTR mutations (5). The non-classic or variant form of CF presents clinical diseases in only a single or subgroup of organs, such as monosymptomatic brochiectasis, chronic pancreatitis, or congenital absence of vas deferens. In general, it is believed that complete loss of CFTR function by the biallelic severe mutations is associated with the classical form, and the reduced CFTR function by mild mutations is associated with the variant form of CF.
Our patient had the classical form of CF presenting both respiratory and gastrointestinal diseases. The chest and abdominal CT scans revealed the bronchiectasis in both lung fields and atrophy of the pancreas, respectively. The patient has suffered from recurrent pneumonia and chronic diarrhea since her infancy. At this admission, her sputum specimens were positive to P. aeruginosa, the most commonly identified pathogen from the respiratory tract cultures of CF patients (13). Finally, CF was confirmed by the positive sweat chloride test.
Currently, over 1,000 mutations and 200 polymorphic loci in the CFTR gene have now been identified (http://www.genet.sickkids.on.ca/cftr/). It is almost impossible to examine all these loci for the clinical diagnosis of CF. Alternatively, genetic tests on the 20 most common loci have been observed that can cover approximately 80-90% of mutations in Caucasian population (Elucigen CF20 Kit, Zeneca Diagnostics, Oxfordshire UK). Therefore, genetic analysis is a useful diagnostic tool when information on the prevalent mutations in the population is available, because identification of the CFTR mutation is highly specific evidence of cystic fibrosis. Unfortunately, the CFTR mutation spectrum in the Korean population has not been fully determined yet.
Previously we have identified the 11 CFTR mutations and polymorphisms circulating in the Korean population (10). Some of them, especially E117G and Q1352H, showed an association with the monosymptomatic bronchiectasis or chronic pancreatitis. Furthermore, molecular study on these mutants showed a reduced Cl- channel activity (10). However, none of the above mutations were identified in the patient's gene of this study. A thorough examination on the coding regions and exon-intron splicing junctions revealed the L441P non-synonymous mutation in one allele. However, mutations in the second allele were not observed. Two explanations are possible. First, the disease causing mutation may exist in the deep intronic regions or may associate with mechanisms not explored in this study, such as gross deletions, complex rearrangements and repeat variations (http://www.hgmd.cf.ac.uk/ac/index.php). In general, most of disease-causing mutations are located on the coding regions and exon-intron splicing junctions where mutations can easily affect the structure and function of the gene product. However, in some cases, mutations in the deep intronic regions also can ablate the gene functions for variable reasons. For example, deep intronic mutations of 1,811+1.6 kbA>G at intron 11 and 3,600+11.5 kbC>G at intron 18 have been shown to be associated with CF in Spanish and African patients, respectively (14, 15). Second, it has been shown that significant proportions of CF patients have only one or no CFTR mutations even after an intensive genetic search (16). Therefore, the defects in other gene may compensate CFTR functions. Especially in the case of variant CF, the defects or environmental factors may contribute to the pathogenesis of CFTR even without second allele mutation in the CFTR gene.
Defects in the protein folding and processing during the secretory pathway of membrane protein are the major molecular pathogenic mechanisms of mutant CFTR (4). This causes an aberrant Cl- and HCO3- transport in the respiratory and pancreatic epithelia (17). The present study is the first study that substantially demonstrated the pathogenicity of the L441P mutation. An integrated molecular physiologic examination revealed that the L441P mutation causes a processing defect. The L441P mutant protein was not fully glycosylated, which implies that the L441P mutant protein can not travel to the Golgi-complex and subsequently to the membrane. This was verified by the absence of L441P mutant protein in the plasma membrane in immunostatining and surface biotinylation experiments. Consequently, the cAMP-activated chloride channel activities of CFTR were completely deteriorated by the L441P mutation.
CF is not common in Asians and there are only a few reports of CF in Korea (6-9). However, analysis of the CFTR haplotype structure revealed that significant proportions of Koreans have minor CFTR mutants, although the major disease causing alleles, such as ΔF508 and G551D, are extremely rare (10). The heterozygote frequency of mild mutations, such as E217G and Q1352H, was estimated to 0.5-1% in the Korean population. This suggests that the incidence of classical and variant CF would be around 1 in 10,000 to 40,000 live births. Although CF is relatively rare in the Korean population, CF should be considered in patients with recurrent chronic respiratory symptoms.




 XML Download
XML Download