

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Endothelial dysfunction is a major early pathologic change and is detected long before the appearance of overt atherosclerotic disease in hypertension. Recent experimental and clinical studies showed acetylcholine, not sodium nitroprusside (SNP), induced vasodilatation is impaired in resistance or conduit arteries from hypertensive patients and animal models. Nitric oxide/endothelial nitric oxide synthase (NO/eNOS) system plays a major role in endothelium dependent vasodilatation along with other vasoactive substances in blood vasculature (1, 2). The impairment of endothelium-dependent dilatation, which is accompanied by the alteration in the expression and/or activity of the endothelial NO synthase, has consistently been observed in conduit arteries and resistance vessels as an effect of hypertension (3, 4).
NO, which is responsible for acetylcholin-induced vasodilation, has been reported to be reduced both in circulation (5), and endothelium (6) in hypertensive subjects. Nitric oxide is synthesized by eNOS, one of a family of heme-containing enzymes that catalyzes the guanidinium moiety of L-arginine to NO and L-citrulline (7). In endothelial cells, eNOS expression and activity are precisely controlled in transcriptional or post-translational levels by protein-protein interactions, Ca2+ concentration and its subcellular distribution (8, 9). Reduced NO generation, without change of eNOS expression, may result from depressed activity of eNOS as a result of endogenous or exogenous inhibitors, reduced substrate L-arginine, decreased cofactor tetrahydrobiopterin (BH4) or decreased bioavailability of NO by increased reactive oxygen species (10, 11).
Caveolae are membrane microdomains, which function as mobile signaling platforms in the plasma membrane, and present ubiquitously in endothelial cells (12). Caveolin-1 is the 22-kDa protein constituent of caveolae, which finely tunes various physiological roles such as activating enzymes as eNOS, modulating signal proteins and mediating transcytosis (13). Caveolin regulates eNOS activity by holding it at scaffold domain in inactive states in vitro. After eNOS is dissociated from caveolin-1 by external stimulation, it forms the complex with calcium/calmodulin or heat shock protein 90. And, then, the eNOS would be activated and increase the NO production (14). Furthermore, the importance of caveolin is increasingly recognized as functioning signalosome and mediator in various diseases such as atherosclerosis, pulmonary hypertension and diabetes (15). In hypertension, caveolin expression is also identified to be increased in endothelial cell and vascular smooth muscle cell and implicated as anti-hypertensive target (16).
3-Hydroxy-3-methylglutaryl Coenzyme-A (HMG-CoA) reductase inhibitors, statins, are the most widely used cholesterol lowering agent, which provide the additional beneficial effects independent of cholesterol lowering. Beneficial effects independent of lipid lowering can also contribute to prevent the atherosclerosis in various populations of patients. The cholesterol-independent or "pleiotropic" effects of statins involve improvement of endothelial function, stability of atherosclerotic plaques, decrease of oxidative stress and inflammation, and inhibition of thrombogenic response (17). Previous studies have demonstrated that statins can stimulate NO release from endothelial cells by eNOS activation. And, the phosphorylation of eNOS may explain, in part, a mechanism of statin-induced eNOS activation and NO release (18).
The aim of this study was to examine whether the HMG-CoA inhibitor, rosuvastatin, alleviates impaired endothelium dependent vasodilatation of the aorta in spontaneous hypertensive rat (SHR) and to investigate the underlying mechanisms involved.
MATERIALS AND METHODS
Animals
Eight week-old SHRs were purchased from SLC, Inc., Japan. The animals were fed with a standard rat chow (CE-2 containing 0.5% NaCl, Purina, Inc., Seoul, Korea) and free access to tap water. SHR were assigned to either a control group (CON, n=11) or a rosuvastatin group (ROS, n=12), rosuvastatin (10 mg/kg/day) was mixed with chow and administered for eight weeks. The amount of rosuvastatin blended with chow was estimated from the weight of chow taken in the previous days. Body weight, serum fasting glucose, cholesterol level, insulin activity were measured. This study was approved by the Animal Care Committee of Seoul National University Bundang Hospital, and followed its recommendation.
Evaluation of vasodilator responses
Vascular relaxations were measured as previously reported (11). In brief, thoracic rings (3-4 mm in length) were suspended in the Krebs solution and oxygenated with 95% O2 and 5% CO2, at 37℃. The rings were contracted maximally with 10-5 M phenylephrine and the endothelin-dependent and -independent relaxations were studied in responses to 109-105 M/L acetylcholine (Ach; Sigma, St. Louis, MO, USA), and to 109-105 M/L sodium nitroprusside (SNP; Sigma), respectively. Endothelium-dependent and -independent relaxation was expressed as the percentage decline of contraction induced by phenylephrine.
Plasma nitrite and nitrate assay
Plasma nitrite and nitrate levels were measured with the Griess method (5). Briefly, the blood samples collected from left ventricular cavity of the rats were added to a microcentrifuge tube containing heparin sodium. The heparin-treated blood samples were centrifuged at 5,000 g, for 10 min, at 4℃ and the plasma was collected. Eighty µL of plasma was mixed with 10 µL of 5 mM β-NADPH, 10 µL (10 U) of nitrate reductase. The samples were allowed to incubate for 1 hr at 37℃ in the dark. Then, 100 µL of Griess reagent (0.1% N-[1-naphthyl] ethylenediamide dihydrochloride, 1% sulfanilamide in 5% phosphoric acid), followed by spectrophotometric measurement at 550 nm. Plasma nitrite and nitrate concentrations in the supernatants were determined by comparison with a sodium nitrite standard curve.
Real time RT-PCR for eNOS expression in aortic tissue
The endothelium and media of aorta was manually separated from adventitia under dissection microscope and homogenized in lysate buffer. And the total RNA was isolated using the RNAqueous filter system (Ambion, Austin, TX, USA). Two µg of total RNA was used to perform real-time PCR. cDNA was synthesized with reverse transcriptase and two µL of cDNA was used for real time PCR. The commercial primer of the sequence for rat eNOS (forward primer: GAA CCT ACA GAG CAG CAA ATC CA; reverse primer: CAG TCC CTC CTG GCT TCC TCC A) and the TaqMan probe (probe: CGA GCC ACA ATC CTG GTC CGT CTT) were used. The reactions were accomplished in triplicate for the samples. The PCR was started at 95℃ for ten minutes followed by 40 two-step cycles of 15 sec at 95℃ and then 1 min at 60℃ by the ABI Prism 7700 Sequence Detection system. Levels of the expression of eNOS as target sequence were quantified by calculating the difference between the target and co-amplified 18S ribosomal RNA.
Western blot analysis for caveolin-1, eNOS and phospho-eNOS protein
Immunoblotting of rat aorta was carried out as previously described (12, 18). The stripped aorta was homogenized and eNOS, phosphorylated eNO and caveolin-1 protein expression was measured in the supernatant of homogenates. SDS/PAGE (12% [w/v] polyacrylamide) was performed. The protein was transferred on to PVDF and incubated with primary polyclonal anti-caveolin-1 antibody (1:1,000; BD, Franklin Lakes, NJ, USA), monoclonal anti-eNOS antibody (1:1,000; Cell Signaling Technology, Inc., Boston, MA, USA) and anti-phospho-eNOS ser 1177 antibody (1:1,000; Cell Signaling Technology, Inc.) overnight at 4℃. The membrane was washed and then incubated with a horseradish peroxidase-conjugated anti-rabbit secondary antibody at room temperature for 1 hr. The immunoreactivity on the membrane was detected using enhanced chemiluminescence (Amersham, Little Chalfont, UK) following the manufacturer's instruction. Normalization for loading differences was accomplished using ratios of the densitometry signals for actin (1:2,000; Calbiochem, Darmstadt, Germany) versus caveolin-1, eNOS or phospho-eNOS protein.
Immunohistochemistry
Tissue preparation was carried out as previously described (16). Briefly, the aortas were fixed with paraformaldehyde in a 0.05 M phosphate buffer (pH 7.4) and cryoprotected with 30% sucrose solution serially and sectioned at 10 µm. The sections were incubated with polyclonal anti-caveolon-1 antibody (1:200 diluted in PBS) for 3 hr at room temperature followed by 1 hr in biotin-conjugated anti-rabbit antibody (Jackson Immunoresearch Lab, West Grove, PA, USA). The sections were then incubated with streptavidin HRP conjugate (Zymed Laboratories, San Francisco, CA, USA) at a 1:150 dilution, and visualized using the aminoethyl carbazole substrate. The sections were then counterstained with Mayer hematoxylin (Sigma).
Statistical analysis
All statistical tests were performed using the SPSS for Windows® (version 15.0, Chicago, IL, USA). Values are expressed as mean±standard error of means. Differences in categorical values between the two groups were analyzed using the chi-square test and differences in continuous variables were analyzed using the Student's t-test. P values of <0.05 were considered statistically significant.
RESULTS
Baseline data
The body weight was similar in each group at the baseline. The change of body weight was higher in rosuvastatin group after 8 weeks, but the difference was not statistically significant (CON vs. ROS; 135.8±15.9 g vs. 148.5±9.9 g, P=NS). The plasma level of total cholesterol (CON vs. ROS; 69.5±4.5 mg/dL vs. 65.7±6.3 mg/dL, P=NS), Triglyceride (CON vs. ROS; 80.7±19.7 mg/dL vs. 70.0±13.7 mg/dL, P=NS) and HDL-cholesterol (CON vs. ROS; 27.9±1.5 mg/dL vs. 27.0±2.2 mg/dL, P=NS) was not different after eight weeks in each group (Table 1). Blood pressure profile after eight week treatment, at fourteen week-age, did not show the statistical difference between each group, even if rosuvastatin-treated group showed the tendency to have lower systolic (CON vs. ROS; 191.9±14.2 mg/dL vs. 186.7±22.1 mg/dL, P=NS) and diastolic blood pressure (CON vs. ROS; 147.0±9.0 mg/dL vs. 144.8±18.2 mg/dL, P=NS) (Table 2).
Endothelium dependent and independent relaxation
The dose-dependent relaxation response to acetylcholine was achieved in both rosuvastatin-treated and control groups with maximal dilatation at maximum concentration (Ach 10-5 M). The relaxation in response to acetylcholine at a dose range of 10-9 to 10-6 M was significantly greater in rosuvastatin-treated group than control group. The peak difference between two groups was seen at Ach 10-8 M. (ROS vs. CON; 82.3±6.6% vs. 73.3±4.8%, P<0.05) However, the maximum relaxation (Ach) between two groups showed no significant difference (Fig. 1A). The response to SNP, a known stimulant for endothelium independent vasodilatation, dose-dependent relaxation response was achieved in both groups without significant differences, which maximum response was at SNP 10-6 M (101.4±2.7% vs. 100.6±3.4%, P=0.32) as well as relaxation to other doses of SNP were similar for the aortic rings of ROS and CON groups (Fig. 1B).
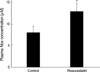
Nitrite and nitrate bioactivity in plasma
Nitrite and nitrate activity in plasma was measured to estimate the nitric oxide bioactivity. The measured plasma nitrite and nitrate was significantly increased in rosuvastatin-treated group compared with control group (CON vs. ROS; 7.6±3.0 µM vs. 12.5±4.5 µM, P<0.05) (Fig. 2).
Expression of eNOS in the aorta
The real time RT-PCR, evaluating the mRNA expression of eNOS in the aorta could not show the difference between rosuvastatin-treated group and control group (Fig. 3A). The protein expression level of eNOS in the aorta was also not different between two groups. However, the level of phosphorylated eNOS, using antibody for Ser1177 of eNOS, was much higher (≈2.3 times) in rosuvastatin-treated group than control group (Fig. 3B).
Down regulation of caveolin-1 by rosuvastatin
The treatment with rosuvastatin (10 mg/kg/day) for 8 weeks resulted in decrease in the protein expression of caveolin-1 in the aorta of SHRs. Immunoblot analysis of aorta extract showed a much lower caveolin-1 expression in the rosuvastatin treated rats than the control group (Fig. 4). Immunostaining of the aorta with anti-caveolin-1 antibody showed a discrete caveolin-1 expression in the endothelial cells. Caveoiln-1 expression was decreased by rosuvastatin treatment, which was demonstrated by the lower expression level in rosuvastatin treated rats compared to control rats (Fig. 5).
DISCUSSION
In this study, we demonstrated that HMG-CoA reductase inhibitor, rosuvastatin, improved the endothelial dysfunction in hypertensive animal without hypercholesterolemia. The hypertension-related endothelial dysfunction seems to occur, at least partly, via a NO-dependent mechanism. Previous studies demonstrated that HMG-CoA reductase inhibitors improved the endothelial dysfunction in hypertensive subjects and its improvement is related to the antioxidant and anti-inflammatory effects irrespective of plasma cholesterol level (19).
The important findings of this study are as follows. First, HMG-CoA reductase inhibitor improved endothelium-dependent vasodilatation of the aorta in the hypertensive animal model, SHR. Second, the improvement of endothelial function by HMG-CoA reductase inhibitor was accompanied by enhanced plasma nitrite and nitrate level reflecting the enhanced production of nitric oxide in endothelium. Third, HMG-CoA reductase inhibitor enhanced the phosphorylation of the eNOS to increase the level of activated eNOS, and subsequently, enhanced the production of nitric oxide without altering the expression level of eNOS. Finally, HMG-CoA reductase inhibitor decreased the expression of caveolin-1 in endothelium of aorta, which could modulate the degree of phosphorylation and, ultimately, enhance the activity of the eNOS.
HMG-CoA reductase inhibitors are widely used in clinical practice and prescribed for the treatment of hypercholesterolemia. Recent clinical and experimental studies have shown that statins have pleiotropic effects, beside modulating lipid, such as anti-inflammatory, anti-proliferative, anti-thrombotic effects, and improving endothelial vasomotor function, all of which may affect cardiovascular outcomes in high risk patients including hypertension (20). In our study, rosuvastatin improved the endothelial dependent vasodilatation in hypertensive rat model without changes of plasma cholesterol level. These results imply that the pleiotropic effect of HMG-CoA reductase inhibitor may play an important role in improving endothelial function unrelated to changes of cholesterol level in hypertension.
Endothelial dysfunction, a precursor to atherosclerotic disease, is favorably affected by HMG-CoA reductase inhibitors. However, the mechanism of the effect of HMG-CoA reductase inhibitors on endothelial function remains unclear. One proposed mechanism for this observation is augmentation of endothelial nitric oxide (NO) bioavailability, resulting in enhanced endothelium-dependent vasodilation. An experimental study demonstrated that HMG-CoA reductase inhibitor improved endothelial dysfunction via reduction of reactive oxygen product and increased expression of eNOS (21). Another evidence for nitric oxide theory is supported from human and animal experimental data showing an increase in endothelial NO production with statin treatment (22). In this study, eight-week treatment of rosuvastatin improved the endothelial function compared with control group (Fig. 1) and increased the nitric oxide metabolite, NOx, in plasma (Fig. 2).
After synthesis and release from the endothelium, nitric oxide rapidly undergoes reaction in the blood to produce molecules, including nitrite, nitrate, nitrosothiol proteins and a-nitrosyl hemoglobin. The plasma nitrite and nitrate concentrations are stable and reliable measure of systemic nitric oxide production and their major source is endothelium (5). In this study, the increased plasma level of nitrite and nitrate indirectly represented the enhanced generation of endogenous nitric oxide in endothelium after treatment of rosuvastatin, which is well correlated to acetylcholine induced endothelium-dependent dilatation (Fig. 1). Furthermore, two groups have been fed with same chow and water for exogenous nitric oxide source under same environment.
The mechanisms of the effect of HMG-CoA reductase inhibitors on nitric oxide bioavailability have not been verified thoroughly. It was demonstrated that HMG-CoA reductase inhibitors could increase nitric oxide production without altering the expression of eNOS in vitro (18). These findings suggest HMG-CoA reductase inhibitors can enhance the nitric oxide production by modulating eNOS activity or other gene products besides regulating the expression of eNOS. The nitric oxide production can also increase by alterations in other molecular system such as caveolin, HSP90 or Cu/Zn superoxide dismutase (14). Recent studies demonstrated selected HMG-CoA reductase inhibitors increased nitric oxide production through HSP90 related mechanism in endothelial angiogenesis model. Treatment with HMG-CoA reductase inhibitor increases eNOS phosphorylation by depressing expression of caveolin-1 or promoting interaction of eNOS with HSP90 or Ca2+/CaM, which makes eNOS activated (18, 23).
The role of eNOS phosphorylation has been elucidated as a determining factor for its own activation, interplaying with its subcellular localization. eNOS is localized to, at least, two subcellular compartments, one of which is the Golgi in perinuclear lesions and the other is the caveolae in the plasma membrane. Membrane bound eNOS is estimated to be more constitutively active and usually phosphorylated by Akt for activation. In addition to the localization in subcellular compartment, binding of eNOS with caveolin-1 inhibits the activation of eNOS and suppresses the nitric oxide production (24). Isolated aortic rings from caveolin-1 null animals were unable to establish a steady-state contractile tone. When acetylcholine was added to the media to stimulate vasorelaxation, the aortic rings from caveolin-1 null mice showed a significantly greater relaxation response than wild-type control rings (25). In SHR, the depressed basal production of bioactive NO was associated with increased expression of eNOS and decreased expression of caveolin-1. At this condition, caveolin-1 played a role as a negative modulator of eNOS to maintain the production of bioactive NO as compensatory mechanism (26).
Several eNOS phosphorylation site, including serine-1177, has important roles in regulating nitric oxide synthesis under various physiological and pathological conditions (14). HMG-CoA reductase inhibitor augments the phosphorylation of eNOS at serine-1177 to increase eNOS activity. Increased phosphorylation at this site by statin is an important mechanism to increase NO production. Ultimately, sustained phosphorylation at serine-1177 is an important factor for the long-term regulation of NO release after statin-stimulated (18). This study demonstrates that HMG-CoA reductase inhibitor increased the eNOS phosphorylation at serine-1177 and enhanced nitric oxide bioavailability without change of eNOS expression accompanied with improved endothelial function.
We demonstrates the rosuvastatin improved endothelium dependent vasodilatation and it was associated with increased nitric oxide bioavailability associated reduced caveolin-1 expression and increased eNOS phosphorylation without changes of eNOS expression in endothelium. These findings suggest that the depressed level of caveolin-1, mediated by HMG-CoA reductase inhibitor, can enhance the phosphorylation and activity of eNOS with decreasing membrane bound form, without change of expression, to increase nitric oxide production. This study revealed the effect of HMG-CoA reductase inhibitor on the endothelial dysfunction via caveolin-1 and eNOS/NO in hypertension, as in other pathologic conditions (27) or in healthy subjects (28).
Baseline characteristics and lipid profiles of two groups were similar. After 8 week treatment of HMG-CoA reductase inhibitor or placebo, the plasma level of total cholesterol was also not different between two groups. Because plasma lipid levels are normally low in rats (in contrast with human beings) and because statins is usually the lack of hypocholesterolemic effect and do not modify lipid profile in rats, they provide an excellent model for studying the other cardiovascular effects (pleiotropic effect) of statins. Our result is in agreement with a recent report in diabetic rats receiving the even higher dosage of rosuvastatin (20 mg/kg/day) without lowering the level of cholesterol (29).
This study has several limitations. First, direct causal relation or proportion of contribution of caveolin-1 with eNOS activation was not proven in this study. In order to prove this linkage, we need caveolin inhibitor, which is not available nowadays, unfortunately. However, recent studies already proved that the caveolin-1 modulate the activation of eNOS in harmony with other signal molecule as Akt, CaMK, PKA, PKC and Hps90 (14). We also demonstrated the increased phosphorylation of Akt in this animal with HMG-CoA reductase inhibitor (data not shown). The contribution of these signal molecules to activity of eNOS would be changed by external stimulation or environmental factors. In this study, even though the exact portion of contribution was not clarified, it is clear that caveolin-1, at least partly, modulates the activity of eNOS under effect of HMG-CoA reductase inhibitor.
Second, rosuvastain treated group showed the trend of lower systolic, diastolic and mean blood pressure without statistical significance in hemodynamic data (Table 2). Some clinical and experimental studies showed that HMG-CoA reductase inhibitor therapy slightly reduced blood pressure. And, furthermore, the changes in blood pressure may influence the endothelial function. As HMG-CoA reductase inhibitor have pleiotropic effects on the vasculature, reduction in blood pressure was accompanied by normalization of endothelial dysfunction, restoration of vasoconstriction to endothelin-1, inhibition of vascular oxidative stress, angiotensin II blocking and upregulation of vascular endothelial NO synthase activity. However, to date, evidence supporting the lowering of blood pressure by HMG-CoA reductase inhibitor is limited to a diminutive number of studies with small sample sizes (30). To elucidate the anti-hypertensive effect of HMG-CoA reductase inhibitor, the more intensive studies with sufficient number of subject should be attempted.
Attenuated endothelium dependent vasodilation of the aorta in SHR is improved by eight week-treatment with HMG-CoA reductase inhibitor. It is at least partly associated with increased NO production accompanied by decreased caveolin-1 expression and altered phosphorylation of eNOS. This result may imply the new therapeutic strategies, regarding caveolin status, to increase endothelial NO generation with HMG-CoA reductase inhibitors in hypertensive subject.






 XML Download
XML Download