

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pulmonary hypertension is a frequent complication of chronic obstructive pulmonary disease (COPD) and associated with a worse survival and increased risk of hospitalization for exacerbation of COPD (1, 2). Pulmonary hypertension is characterized by thickening of pulmonary artery walls, narrowing of pulmonary artery lumina, and elevation of pulmonary vascular resistance. These pathophysiologic findings could be explained by the effects of hypoxemia, shear stress, endothelial dysfunction, and inflammatory reaction (3). Alveolar hypoxia may play a role in the pathogenesis of pulmonary hypertension associated with COPD. However, structural and functional changes in the pulmonary arteries have also been observed in normoxic patients with mild-to-moderate COPD (4, 5).
Several studies (6, 7) suggested that chronic inflammation contribute to pulmonary vascular remodeling. C-reactive protein (CRP) has both proinflammatory and antiinflammatory actions. Proinflammatory effects include the induction in monocytes of inflammatory cytokines and tissue factors (8, 9). In pulmonary hypertension with COPD patients, the level of CRP correlate with pulmonary artery pressure and is an independent predictor of systolic pulmonary artery pressure (6).
Endothelin-1 (ET-1) is a potent vasoconstrictor peptide produced by the vascular endothelium (10). Increased production of ET-1 in vascular endothelium may result in remodeling of the pulmonary vasculature, leading to the development of pulmonary hypertension in patient with COPD. ET-1 levels are elevated in patient with pulmonary hypertension and strongly correlated with disease severity (11, 12). However, Bacakoğlu et al. (13) reported that plasma levels of ET-1 did not significantly differ between COPD patients with and without pulmonary hypertension. To our knowledge, there are few reports available concerning the relationships between the plasma level of CRP and ET-1 in patients with COPD. Therefore, the purpose of this study was designed to investigate the degree of plasma CRP and ET-1 levels in COPD patient with and without pulmonary hypertension. In addition, we sought to determine whether plasma levels of ET-1 related to that of CRP in patients with COPD.
MATERIALS AND METHODS
Subjects
Patient with a diagnosis of COPD, determined according to the American Thoracic Society (ATS) guidelines (14), were recruited from January to December 2009 in the study. The inclusion criteria were a history of smoking, an FEV1 (forced expiratory volume in 1 sec)/FVC (forced vital capacity) ratio <70%, and an FEV1 <80% of the predicted values. Exclusion criteria were respiratory disorders other than COPD, pulmonary embolism, infectious diseases, malignancy, left ventricular dysfunction, recent surgery, and severe endocrine, hepatic, or renal diseases. At recruitment, height and weight were measured and the body mass index was calculated (weight [kg]/height [m2]).
Pulmonary function test
All pulmonary function tests were evaluated using a Sensormedics 2400 Unit (Sensormedics; San Diego, CA, USA) that met the standards of the ATS (14). Most pulmonary function tests were obtained on the same day as the echocardiography and/or laboratory investigations. Three technically acceptable measurements were performed in each patient, and highest one was included to the analyses. Acceptable maneuvers were defined as those with sharp peak in the flow curve and an expiratory duration greater than six seconds. FEV1, FVC, and lung volume were expressed as percentage of predicted values.
Echocardiography
Echocardiography was performed using a commercially available machine (Siemens Medical Solution; Malvern, PA, USA) equipped with a 2.5 MHz probe. Tricuspid regurgitation flow was identified by color flow Doppler techniques, and the maximal jet velocity was recorded from the parasternal or apical window with the continuous wave Doppler echocardiographic probe. The trans-tricuspid pressure gradient (ΔP) can be calculated from the maximum velocity of tricuspid regurgitant jets (Vmax) and the simplified form of the Bernouilli equation: ΔP(mmHg)=Vmax(m/s)×4. An estimation of pulmonary artery pressure is obtained by adding an estimate of right atrial pressure to ΔP. Pulmonary hypertension was generally defined as a mean pulmonary artery pressure (Ppa) greater than 25-35 mmHg with a normal pulmonary artery wedge pressure (15). However, the symposium on pulmonary hypertension suggested that a systolic pulmonary artery pressure of 35 mmHg represents the cut-off value for pulmonary hypertension when used by Doppler echocardiography (16). Therefore, we have divided patients into those without pulmonary hypertension (systolic Ppa <35 mmHg) and those with pulmonary hypertension (systolic Ppa ≥35 mmHg).
C-Reactive protein
Venous blood samples were drawn into empty tubes and serum was obtained by centrifuging at 3,000 rpm for 15 min. The samples were stored at -70℃ until analyzed. Serum high sensitive CRP was measured by the high sensitivity nephelometric method (Dade Behring; Marburg, Germany). The results were given as mg/L.
Endothelin-1
Peripheral venous blood samples were obtained from antecubital vein in the patients. Serum was prepared by drawing 5 mL of blood into tubes containing EDTA (Becton Dickinson; Plymouth, UK). The plasma was obtained by centrifuging 3,000 rpm 15 min. The supernatants were stored at -70℃ until analyzed. ET-1 plasma level was measured by a commercially available sandwich enzyme-linked immunoassay (R&D systems; MN, USA). The results were given as pg/mL.
Statistical analysis
A statistical software package (SPSS, version 17.0; SPSS Inc; Chicago, IL, USA) was used for all statistical analyses. The data are reported as mean±SD for variables that were normally distributed. The data were expressed as the median and interquartile range (IQR) for variables that were not normally distributed. Differences between the groups (patients with vs without pulmonary hypertension) were analyzed using unpaired Student's t test and Mann-Whitney U test. Correlation coefficients were calculated with the Spearman rank test. A P value of <0.05 was considered statistically significant.
RESULTS
Clinical characteristics of the study population
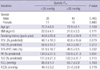
The 108 patients with COPD (82 male and 26 female) participated in this study. Forty-one patients were classified as having moderate COPD (FEV1 ≥50%), 55 patients as severe COPD (30% ≤FEV1 <50%), 12 patients as very severe COPD (FEV1 <30%) when stratified by ATS guidelines (15). The clinical characteristics of the two groups are shown in Table 1. Pulmonary hypertension was present in 58 patients (mean systolic Ppa, 49.4±12.9 mmHg) and was absent in 50 patients (mean systolic Ppa, 23.6±4.3 mmHg). The two groups of subjects were similar with regard to age, sex, smoking history (packs/year) and body mass index. Compared to patients without pulmonary hypertension, patient with pulmonary hypertension had a trend toward lower FEV1 and higher residual volume. However, it did not reach statistical significance. Also, no differences were observed in blood gas analysis results between the two groups.
CRP and ET-1 levels in patients with and without pulmonary hypertension
Plasma CRP levels were significantly higher in patients with pulmonary hypertension than in patients without hypertension (median, 8.0 mg/L [IQR, 3.0-13.4 mg/L] vs. median, 4.4 mg/L [IQR, 2.9-7.5 mg/L]; P=0.026) (Fig. 1). Plasma ET-1 levels were significantly higher in patients with pulmonary hypertension than in patients without hypertension (median, 4.1 pg/mL [IQR, 1.9-6.6 pg/mL] vs. median, 1.7 pg/mL [IQR, 1.1-3.7 pg/mL]; P=0.001) (Fig. 2). There were significant positive correlations between the plasma ET-1 level and CRP level in patients with COPD (without or with pulmonary hypertension) (r=0.712, P=0.001) (Fig. 3). For COPD patients, systolic Ppa correlated significantly with plasma CRP levels (r=0.461, P=0.001) and plasma ET-1 levels (r=0.531, P=0.001) (Fig. 4, respectively).
DISCUSSION
The present study demonstrated that COPD patients with pulmonary hypertension had higher plasma CRP and ET-1 levels compared to those without pulmonary hypertension. In patients with COPD, plasma CRP and ET-1 levels correlated significantly with systolic pulmonary artery pressure. In addition, there were significant positive correlations between the plasma ET-1 level and CRP level in patients with COPD. These findings suggest that CRP and ET-1 play a role in the pathogenesis of pulmonary hypertension associated COPD.
Cigarette smoke in rats induced proliferation of cells in pulmonary vasculature and in the airways (17). Airway inflammation, induced by chronic cigarette smoking, may lead to pulmonary vascular changes. The primary regulators of CRP are interleukin (IL)-6, IL-1b and a tumor necrosis factor (TNF)-α, which are secreted by neutrophil granulocytes and macrophages at sites of injury. Inflammatory cytokines such as IL-6 and TNF-α are markers of elevated pulmonary artery pressure in COPD (6, 7). A recent study demonstrated that CRP levels correlated with pack-years of smoking and predicted all-cause mortality in patients with mild to moderate COPD (18). In COPD patients with pulmonary hypertension, serum CRP level was higher compared to those without pulmonary hypertension (6). Our data demonstrated that plasma CRP levels were significantly higher in patient with pulmonary hypertension compared to those without hypertension. In addition, we found positive correlations between plasma levels of CRP and systolic pulmonary arterial pressure. These results suggested that systemic inflammation might be related to pulmonary hypertension in patient with COPD. However, a recent study in patients with moderate to very severe COPD showed that there was no relationship between CRP levels and likelihood of survival (19). Further long-term studies are needed to address the physiological role and clinical outcome of CRP in COPD patients with pulmonary hypertension.
ET-1 is a vasoactive mediator that causes vasoconstriction and vascular wall proliferation. Therefore, ET-1 has been suggested that ET-1 may contribute to increases in pulmonary arterial tone or smooth muscle proliferation in patients with pulmonary hypertension. Immunocytochemical analysis of lung specimen demonstrated that pulmonary hypertension was associated with the increased expression of ET-1 in vascular endothelial cells (20). Furthermore, there is a significant correlation between pulmonary ET-1 concentration and pulmonary vascular resistance (21). Our findings confirm these observations (20, 21) by showing that plasma ET-1 level was significantly higher in patient with pulmonary hypertension than in patients without hypertension. In addition, a significant relationship was observed between ET-1 level and systolic pulmonary artery pressure. These evidences suggest that ET-1 may result in remodeling of the pulmonary vasculature, leading to the development of pulmonary hypertension in patients with COPD. However, our results are based on single measurements of CRP, ET-1 and pulmonary artery pressure, which may not reflect these relationships over time. Therefore, this study does not provide information about the time-course relationship between CRP and ET-1 and pulmonary hypertension. Several reports of ET-1 levels in COPD patients with pulmonary hypertension have been conflicting (13, 22). Fujii et al. (22) showed that there was no correlation between the plasma ET-1 level and pulmonary artery pressure. Further studies are necessary to elucidate these conflicting results.
ET-1 is also thought to have a proinflammatory effect in the airways, being both a chemoattractant and upregulator of other inflammatory mediators, such as IL-6 and IL-8 (25). Subsequently, IL-6 and IL-8 stimulate the production of CRP in the liver. ET-1 levels are found elevated in several pulmonary disease such as asthma (24, 25), COPD (22), and idiopathic pulmonary fibrosis (26). These pulmonary diseases are associated with inflammatory response, characterized by increased CRP levels. Several studies (27, 28) have shown that both ET-1 and CRP are implicated in the inflammatory process of cardiovascular diseases. Wang et al. (27) demonstrated that ET-1 induces CPR production in rat vascular smooth muscle cells. On the other hand, Gemici et al. (29) showed that there was no correlation between the plasma levels of ET-1 and CRP in coronary artery disease. However, there are few reports available concerning the relationships between the plasma level of CRP and ET-1 in patients with COPD. We found out that there was a significant positive correlation between plasma ET-1 and CRP levels in the COPD patients. This finding supports a concept of the link between plasma CRP and ET-1 in patients with COPD.
In conclusions, our study demonstrated that pulmonary hypertension in patients with COPD were associated with higher plasma CRP and ET-1 level. In addition, plasma levels of CRP and ET-1 were significantly correlated with systolic pulmonary artery pressure in patient with COPD. These findings support a possibility that CRP and ET-1 correlate to pulmonary hypertension in COPD patients. Further investigations are necessary to reveal the pathophysiological role of systemic inflammation in the pulmonary hypertension associated with COPD patients.




 XML Download
XML Download