

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital central hypoventilation syndrome (CCHS) is a rare autosomal dominant disorder of the autonomic nervous system (ANS) characterized by an abnormal autonomic ventilatory response to progressive hypercapnia and sustained hypoxemia. Most patients present with hypoventilation or apnea during the neonatal period, typically while asleep and/or awake without any other associated diseases such as cardiac, pulmonary, neuromuscular or brain stem abnormalities (1, 2). A rare association with Hirschsprung disease (Haddad syndrome) was first described in 1978; five cases of Haddad syndrome have been reported in Korea (3-7). It is because of the association with aganglionosis of the bowel that a number of candidate genes have been considered.
A common pathogenesis involving neural crest-derived cell lineages has been suggested (8). Studies of genes pertinent to the early embryologic development of the ANS include the mammalian achaetescute homolog-1 (MASH1), bone morphogenic protein-2 (BMP2), engrailed-1 (EN1), TLX3, endothelin converting enzyme-1 (ECE1), endothelin-1 (EDN1), PHOX2A, and PHOX2B among 67 probands with CCHS. No disease-defining mutations were identified in MASH1, BMP2, EN1, TLX3, ECE1, EDN1, or PHOX2A. However, 97% of patients with CCHS have been found to be heterozygous for the exon 3 polyalanine expansion mutation identified previously in PHOX2B (9). PHOX2B has been mapped to chromosome 4p12 and consists of 3 exons. It encodes a highly conserved paired-like homeo box transcription factor of 314 amino acids linked to the RET-GDNF signaling pathway. The DNA binding homeo domain is encoded by exon 2, whereas exon 3 encodes two short and stable polyalanine tracts of 9 and 20 residues (2). Expression studies performed in mice and humans have shown that this is a master regulatory gene, crucial for the normal development of the peripheral and central ANS (10-12). We herein report a case of CCHS in a Korean patient that was confirmed during the neonatal period by the identification of PHOX2B mutations.
CASE REPORT
Clinical history
A 3,070 g male at 41 weeks gestation age was born in our hospital via cesarean section because of cephalopelvic disproportion on July 26, 2007. His mother was healthy during her pregnancy (gravida 1, para 0). The boy was well at birth, with Apgar scores of 7 and 9 at 1 and 5 min, respectively. He repeatedly had apnea with cyanosis and desaturation, noticed from the 15 hr after birth, and improved by awakening. He sometimes developed seizures when the PaCO2 accumulated, which needed supportive care with mechanical ventilation. No abnormalities were noted on the chest radiograph, EEG (Fig. 1), MRI of the brain, echocardiography and screening blood tests for metabolic disease. Symptoms of respiratory failure with slow and irregular respiratory effort, cyanosis and seizures due to hypercapnia appeared during sleep but not during wakefulness (Fig. 2). The diagnosis of CCHS was suspected. Peripheral blood samples for the gene study were obtained from the patient and his mother at the age of 33 days with consented document of the family.
The infant could not weaned from mechanical ventilation due to sleep apnea. He had a tracheostomy and was discharged with a home mechanical ventilation. At the age of 27 months, he is healthy except still ventilator-dependent during sleep. The patient achieved normal growth and development.
DNA preparation
Blood (4 mL) was collected into an EDTA tube from the patient and his mother. Genomic DNA was obtained using a Puregene reagent kit (Gentra, Minneapolis, MN, USA) according to the manufacturer's instructions.
Genotyping of PHOX2B polyalanine repeat sequence
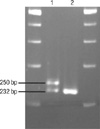
The PHOX2B exon 3 region coding for the polyalanine repeat was amplified with primer pair 5'-CCAGGTCCCAATCCCAAC-3' (forward) and 5'-GAGCCCAGCCTTGTCCAG-3' (reverse) (Fig. 3). The PCR reactions were carried out using 0.25 U AmpliTaq Gold polymerase (Applied Biosystems, Foster City, CA, USA) in a total volume of 25 µL, containing 50 ng genomic DNA, 0.3 µM primers, 2.5 mM MgCl2, and 0.2 mM dNTPs. The amplification was performed with an initial denaturation at 95℃ for 10 min followed by 35 cycles of denaturation at 94℃ for 30 sec, annealing at 57℃ for 30 sec, and extension at 72℃ for 30 sec. Final extension was at 72℃ for 10 min. After electrophoresis of the PCR products on a 4% denaturing polyacrylamide gel, the allele repeat number was determined by comparison of bands to known size standards (232 bp for normal 20 repeat allele) (9).
Sequence analysis of PHOX2B exons 2 and 3 region
PHOX2B exons 2 and 3 were amplified with primer pairs noted in Table 1. The amplification of exon 2 was carried out using 1.25 U AmpliTaq Gold polymerase (Applied Biosystems) in a total volume of 25 µL, containing 50 ng of genomic DNA, 0.5 µM of primers, 1 mM MgCl2, and 0.2 mM dNTPs. The amplification of exon 3 was performed using the GC-RICH system (Roche Molecular Biochemicals, Indianapolis, IN, USA) for its high GC content. The amplification for both exons was performed with an initial denaturation at 95℃ for 8 min, followed by 35 cycles of denaturation at 95℃ for 1 min, annealing at 62℃ for 1 min, and extension at 72℃ for 45 sec. Final extension was at 72℃ for 10 min. The PCR products were column-purified and sequenced on an Applied Biosystems 3130 genetic analyzers (Applied Biosystems).
Results of mutation
A polyalanine repeat expansion mutation was identified in the baby with CCHS. He carried a normal allele, as well as the expanded alleles (Fig. 4). The expanded alleles contained insertions of 18 bp DNAs for coding 6 alanines. An extra band with a size >232 bp was observed in the patient, indicating expansion of the polyalanine track. His mother had normal alleles (Fig. 4).
DISCUSSION
CCHS was suspected in the present case based on following findings: 1) hypoventilation during sleep with no increase of respiratory frequency following severe desaturation, 2) onset of symptoms during the first day of life and a history of recurrent hypercapnic respiratory failure, and 3) absence of primary neuromuscular, lung or cardiac disease, or an identifiable brainstem lesion.
CCHS occurs in association with Hirschsprung disease (Haddad syndrome) by 15-20% frequency, tumors of neural crest origin (neuroblastoma, ganglioneuroblastoma, glanglioneuroma) by 3%, and growth hormone deficiency by <1% (13). Korean patients with CCHS combined with Hirschprung disease have been reported based on clinical symptoms without genetic analysis (3-7). The occurrence of CCHS and Hirschsprung disease suggests a common molecular pathogenesis involving defects of one or more genes that control the development of neural crest-derived cell lineages. This patient did not have any other abnormalities but will be monitored for a malignancy of the ANS in the future.
The clinical outcome of CCHS is variable. Infants with CCHS may present with hypoventilation of variable severity, ranging from complete apnea during sleep, severe hypoventilation during wakefulness, to mild hypoventilation during sleep (9, 14). The patient reported here, required a tracheostomy and used a home mechanical ventilator in the pressure-support mode. The patient became ambulatory while awake and intermittently required ventilator support during sleep only.
The role of genes in the pathogenesis of CCHS has been widely investigated. Several studies have shown that a heterozygous mutation in PHOX2B is sufficient to cause CCHS, and this has confirmed its autosomal dominant inheritance pattern with incomplete penetrance (9, 10, 15-19). This gene is known to code for a highly conserved transcription factor and plays a key role in the development of ANS reflex circuits in mice, hence the association with CCHS. Mapping of the PHOX2B mutation in chromosome 4p12 has been detected in approximately 97% of patients with CCHS, and the majority of expanded alleles contained 25-27 polyalanine repeats (9). This patient showed the same PHOX2B mutation with expanded alleles containing 20 polyalanine repeats. Weese-Mayer et al. (9) suggested that the length of the PHOX2B polyalanine repeat mutation is associated with number of ANS symptoms and that there is also a significant association between the number of repeats in the mutations and the ventilation support required. The current patient had a relatively small number of polyalanine repeats compared to previous cases and required intermittent ventilator support during sleep, which was for less than 12 hr a day.
Clinically, evaluation of the PHOX2B expansion could be used as a predictive test for CCHS, therefore it might be particularly useful for parents of a child with CCHS and grown children with CCHS. In addition, prenatal testing can then be used to predict the severity of affected individuals in subsequent pregnancies. The identification of mutations in PHOX2B is important for the differential diagnosis in children with confusing symptoms such as asphyxia, prematurity, bronchopulmonary dysplasia, and neonatal seizures, and may lead to improved treatment options for children with CCHS (9). This is the first report of CCHS confirmed by a PHOX2B mutation in Korea.




 XML Download
XML Download