

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Congenital nephrotic syndrome (CNS) is defined as nephrotic syndrome which manifests in utero or during the first 3 months of life (1). Although CNS may develop secondary to underlying diseases such as various kinds of congenital infections, most of CNS is primary lesion of genetic origin. Recent genetic studies disclosed four major causative genes implicating CNS; the NPHS1 gene encoding nephrin, the NPHS2 gene encoding podocin, the WT1 gene encoding the nuclear transcriptional factor Wilms tumor suppressor gene 1, and the LAMB2 gene encoding laminin β2 chain (2).
CNS of Finnish type (CNF, OMIM #602716) is an autosomal recessive disease due to NPHS1 mutations. CNF is the most common CNS in Finland with the incidence of 1 per 10,000 births. However, patients with CNF have also been reported worldwide. Most of the patients develop nephrotic syndrome at birth or soon after birth, which progresses rapidly to end-stage renal disease. Kidney transplantation is the only curative treatment modality currently (3-5).
CASE REPORTS
Case 1
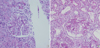
A 2 months old male infant was admitted to a local hospital due to generalized edema. On prenatal examination, high α-fetoprotein (AFP) level (20.85 MoM) in the maternal serum was detected at the 18th gestational week. The baby was born at 36 weeks of gestational age with a birth weight of 2.57 kg. The weight of placenta was reported to be normal. Apgar scores at 1 and 5 min were 9 and 10 points, respectively. However, he showed shortness of breath and abdominal distension soon after the birth. He has normal male external genitalia. Laboratory tests revealed markedly hypoalbuminemia (serum albumin 1.2 g/dL), massive proteinuria (urinary protein excretion 7,959 mg/m2/day), and hypothyroidism. There was no evidence of congenital infection. The parents and his older brother are all healthy except that his mother is a hepatitis B virus antigen carrier. Captopril (0.1 mg/kg/day) and synthyroid (2.7 µg/kg/day) were started as well as intermittent albumin infusions. However, generalized edema was aggravated with frequent need of albumin infusion. At 2 months of age, he was transferred to Seoul National University Children's Hospital because of intractable generalized edema and respiratory difficulty. On admission, his body weight was 4.49 kg (3-10 percentile), and height was 52 cm (<3 percentile). He showed mild chest retraction, and breathing sound was coarse with crackles on both lung fields. The serum albumin was 1.6 g/dL, serum creatinine was 0.3 mg/dL, random urine protein/creatinine ratio was 110 mg/mg, platelet count was 888×103/µL, and serum immunoglobulin G level was 64 mg/dL (normal range, 176-201 mg/dL). The nasopharyngeal aspirate was positive for human respiratory syncytial virus A. Thus, in addition to supportive lung care, intravenous immune globulin (0.5 g/kg) was given. He was medicated with indomethacin (4 mg/kg/day), captopril (4.7 mg/kg/day), dipyridamole (3 mg/kg/day), warfarin, and synthyroid as well as high protein (up to 4 mg/kg/day) and high calorie diet (120 Cal/kg/day). However, frequent albumin infusion (2 g/kg/day) had to be continued to control respiratory problem and generalized edema, and a Chemoport® was implanted into right atrium via right external jugular vein to secure a central venous line. An open renal biopsy was performed at 4 months of age, which showed diffuse mesangial proliferation (Fig. 1A, 2A). Genetic analysis of the NPHS1 gene revealed compound heterozygous frame-shifting mutations, 8-bp deletion in exon 16 (c.2156_2163 delTGCACTGC causing p.L719DfsX4), and 1-bp insertion in exon 24 (c.3250_3251insG causing p.V1084-GfsX12) (Fig. 3A, B). The former mutation was inherited from mother, and the latter from father. During follow-up, he was admitted 2 more times due to central line infections. Thus, at 8 months of age, Chemoport® removal and unilateral nephrectomy were done to control the recurrent infections and to reduce the degree of proteinuria and the need of albumin infusions, respectively.
Case 2
A female newborn baby was admitted to neonatal intensive care unit of Kyung Hee University Medical Center due to respiratory difficulty, which was detected at birth. She was born at 38 weeks of gestational age, and the birth weight was 2,920 gm. Apgar scores at 1 and 5 min were 7 and 9 points, respectively. There was no abnormality on routine prenatal examinations. She is the first baby of healthy parents. On physical examination, she showed tachypnea and generalized edema. The serum albumin was 1.5 g/dL, serum creatinine was 0.3 mg/dL, random urine protein/creatinine ratio was 64.4 mg/mg, platelet count was 377×103/µL, and serum immunoglobulin G level was 146 mg/dL. She has normal female external genitalia. Captopril (1.5 mg/kg/day), dipyridamole and synthyroid were started as well as daily intravenous albumin infusions (1 g/kg/day). A kidney biopsy, which was done at 1 month of age, revealed diffuse mesangial proliferation (Fig. 1B, 2B). Genetic analysis of the NPHS1 gene revealed compound heterozygote mutations, a missense mutation in exon 11 (c.1381G>A causing p.R460Q) inherited from mother and a nonsense mutation in exon 18 (c.2442C>G causing p.Y814X) inherited from father (Fig. 3C, D). At 1.5 months of age, she was discharged, and her family emigrated to Japan.
DISCUSSION
The cardinal clinical manifestation of nephrotic syndrome is massive proteinuria due to impairments of the glomerular filtration barrier, which is composed of 3 layers: the fenestrated endothelium, the glomerular basement membrane, and the podocyte foot processes with bridging slit diaphragms. Recent molecular genetic studies have identified several genes involved in the pathogenesis of hereditary nephrotic syndromes, and most of these genes encode proteins of the glomerular podocyte or the glomerular basement membrane or nuclear transcription factors expressed in the podocytes (NPHS1, NPHS2, CD2AP, ACTN4, TRPC6, PLCE1, LAMB2, WT1, etc).
The onset of nephrotic syndromes associated with genetic defects differs from each other. The NPHS1 gene, which encodes nephrin, a major component of the slit diaphragm, is the first gene identified to be associated with hereditary nephrotic syndrome, CNF (3). Mutations in NPHS1 are exclusively detected in patients with CNS (3). Podocin, which is encoded by the NPHS2 gene, is another essential component of the slit diaphragm and a close interactor of nephrin. Genetic defect of podocin is the cause of early-onset autosomal recessive steroid-resistant nephrotic syndrome (OMIM #600995) (8). The WT1 gene functions both as a tumor suppressor and a critical regulator of kidney and gonad development. Germline mutations in WT1 result in two clinical syndromes with glomerular diseases, Denys-Drash syndrome (OMIM #194080) and Frasier syndrome (OMIM #136680), as well as isolated steroid-resistant nephrotic syndrome (9, 10). The LAMB2 gene encodes laminin-β2 chain, which is one of the major components of the glomerular basement membrane. Mutations in LAMB2 present with a spectrum of clinical phenotypes from isolated early onset nephrotic syndrome to the full syndromic phenotype of Pierson syndrome with CNS, diffuse mesangial sclerosis, microcoria, and mental retardation (OMIM #609049) (11-13). Mutations in PLCE1, which is also called as NPHS3 and encodes phospholipase C epsilon, can cause autosomal recessive early onset nephrotic syndrome with rapid progression to end-stage renal disease (14). Mutations of the ACTN4 (encoding α-actinin-4), TRPC6 (encoding canonical transient receptor potential 6 ion channel) and CD2AP (encoding CD2-associated protein) genes are primarily associated with autosomal dominant focal segmental glomerulosclerosis in adults (15-17).
A recent genetic study of a large number of European and Turkish children with CNS disclosed that 84.8% of patients with CNS (39 of 46 families) carried disease-causing mutations in one of the four genes; 18 (39.1%) in NPHS1, 18 (39.1%) in NPHS2, 1 (2.2%) in WT1, and 2 (4.4%) in LAMB2 (2). In our country, the incidence of WT1 mutations in children with steroid-resistant nephrotic syndrome is similar to those of other countries (18), and cases with LAMB2 mutations have also been reported (13). In contrast, NPHS2 mutations appear to be very uncommon in Korea as same as in other Far East Asian countries (China and Japan) (18). However, there has been no genetic study of CNS with NPHS1 mutations before this paper in Korea.
Precise diagnosis of CNF can be confirmed only by genetic analysis of NPHS1. To date, about 100 different mutations have been identified in the NPHS1 gene. Two founder mutations, Finmajor (c.121delCT causing p.L41fsX90) and Finminor (p.R1109X), account for nearly 90% all affected Finnish patients (4, 5). On the other hand, most patients outside Finland have individual mutations of different types (2, 4, 14, 19). Among the four mutations detected in this study, one nonsense mutation (p.Y814X) is novel, and other three are known mutation (3, 20).
Compared with many other genetic disorders, NPHS1 shows relatively little phenotypic variation (21). Most of the patients with CNF are born prematurely with abnormally enlarged placenta (the placental weight is over 25% of the newborn weight). Proteinuria begins in utero and and full-blown nephrotic syndrome follows soon after birth. The clinical courses are commonly complicated by poor nutritional status, growth delay, recurrent infections, thromboembolism, and hypothyroidism, and progression to end-stage renal disease within 3-8 yr of life is inevitable (5).
Renal biopsy does not reveal the etiology of CNS, because the pathologic findings overlap in different genetic entities. The most characteristic renal pathologic findings of CNF are expansion of glomerular mesangium and dilations of the proximal and distal tubules. Effacement of podocyte foot processes and disappearance of the filamentous image of podocyte slit diaphragm are seen in electron microscopy, although none of these findings are pathognomonic for CNF (5, 21). However, immunohistochemistry for nephrin expression in a biopsy sample is useful.
Steroids or other immunosuppressive drugs are not effective at all in all forms of hereditary proteinuric diseases including CNF, although rare exceptional cases have been reported (14). Thus, successful kidney transplantation is the only curative treatment. During infantile period with active nephrotic syndrome, several conservative managements are recommended to control severe edema such as daily or every other day albumin infusion with intermittent gamma globulin replacement, and low-salt diet. A combination treatment of angiotensin converting enzyme inhibitor and indomethacin lowers intraglomerular pressure and, therefore, decreases urinary protein excretion. It is also important to prevent and treat infectious and thrombotic complications. Optimal nutrition including vitamin and thyroid hormone should also be provided for the patients to grow and develop as normally as possible. However, unilateral or bilateral nephrectomy and early dialysis before the development of renal failure may be required in some patients to control massive protein loss. When the patient reaches a body weight of 8 to 9 kg, renal transplantation can be considered (1, 5).
Elevated AFP levels in amniotic fluid and maternal serum, which reflects fetal proteinuria in utero, can be used as a prenatal diagnostic marker of CNF. However, because heterozygous fetal carriers of NPHS1 mutations may have temporarily elevated AFP levels, prenatal diagnosis should be based on genetic testing whenever possible in high risk families. Otherwise, repeated measurement of amniotic fluid AFP before the 20th week of pregnancy is recommended (5). In case of no family history or if the mutations in the affected sibling(s) were not identified yet, prenatal diagnosis of CNF may be based on high amniotic fluid AFP level only, because whole sequencing the NPHS1 (total 29 exons) gene is time consuming (5). Amniotic fluid AFP level can be elevated fetal anencephaly or other malformations as well. Elevated AFP levels in amniotic in maternal serum was detected in Case 1 of this study, but nevertheless the prenatal diagnosis of CNF was missed. It may be due to unfamiliarity of CNF to clinicians in our country.
In conclusion, this is the first report of Korean children with CNF, which was confirmed by molecular genetic studies. Precise genetic diagnosis is important for proper management of the patients and accurate genetic counseling of the families. In addition to NPHS1, further molecular genetic study of NPHS2, WT1, and LAMB2 will provide more detailed information about the genetic background in the Korean patients with CNS.


 XML Download
XML Download