

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
In the renin-angiotensin-aldosterone systems, angiotensin II is well known to mediate progressive renal disease not only by elevating glomerular pressure, but also by non-hemodynamic effects on the kidney, such as proliferation of renal cells including mesangial cells, recruitment of mononuclear cells into the kidney by NF-κB-mediated transcription of chemokines, and stimulating renal TGF-β, fibronectin and type I collagen expression (1, 2). Angiotensin converting enzyme inhibitors and angiotensin receptor blockers have shown efficacy in slowing the progression of diabetic and non-diabetic renal disease as well as in decreasing proteinuria (1).
Recently, emerging and convincing evidences have shown the independent role of aldosterone in the progression of renal disease. In the remnant kidney model using rat, aldosterone induced proteinuria, hypertension and glomerulosclerosis (3). In addition, inhibition of aldosterone by spironolactone not only slowed the development of glomerulosclerosis, but also induced the regression of existing glomerulosclerosis in the same model (4). Aldosterone infusion developed malignant nephrosclerosis in saline-drinking stroke-prone hypertensive rats, independent of the effects of blood pressure (5) and induced severe inflammation and fibrosis by macrophage infiltration and cytokine up-regulation in saline-drinking rats (6).
The role of TGF-β in the aldosterone-induced renal injury has been suggested in animal models of various renal diseases. In a rat model of chronic cyclosporine A (CsA) nephrotoxicity, spironolactone treatment reduced arteriolopathy and tubulointerstitial fibrosis by prevention of CsA-induced up-regulation of TGF-β, collagen I and fibronectin (7), and also prevented the progression of renal injury in preexisting chronic CsA nephrotoxicity (8). Aldosterone also played a role in diabetic renal injury, which was demonstrated by increasing glomerular and tubulointerstitial macrophage infiltration, plasminogen activator inhibitor-1 and TGF-β expression in streptozotocin-induced diabetic rats (9), and by increasing renal tissue and urinary levels of monocyte chemoattractant protein-1 as well as renal macrophage infiltration in type 2 diabetic rats (10). Juknevicius and colleagues demonstrated that aldosterone infusion for 3 days in normal rats increased urinary TGF-β excretion without changes in blood pressure or evidence of kidney damage (11).
The renal protective effects of inhibiting aldosterone were also demonstrated in chronic kidney disease patients. In a prospective randomized open-label study, spironolactone treatment for 1 yr in patients with chronic kidney disease already treated with angiotensin-converting enzyme inhibitors and/or angiotensin type 1 receptor antagonists significantly decreased proteinuria without any change in glomerular filtration rate (12). Similar results were observed in another clinical study (13).
Putting these results from various experimental studies together, it is positive that aldosterone stimulates renal TGF-β expression in the course of progressive renal injury. Recently, Han et al. (10) described the aldosterone stimulated NF-κB DNA binding activity and subsequently monocyte chemoattractant protein-1 expression in mesangial cells. However, it has not been clearly determined if aldosterone-induced TGF-β expression was mediated through the extracellular-signal regulated-kinase 1 and 2 (ERK1/2), c-Jun N-terminal kinase (JNK) and activator protein-1 (AP-1) pathways in mesangial cells. In this study, we examined the role of ERK1/2, JNK and activator protein-1 in the intracellular signaling pathways of aldosterone-induced TGF-β1 expression in rat mesangial cells.
MATERIALS AND METHODS
Reagents
Recombinant human TGF-β1 and anti-human TGF-β1 antibodies were purchased from R&D Systems (Minneapolis, MN, U.S.A.). Anti-c-Jun antibody, anti-c-Fos antibody and AP-1 consensus oligonucleotides (5'-CGC TTG ATG ACT CAG CCG GAA-3') were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, U.S.A.). JNK and ERK1/2 antibodies were purchased from Cell Signaling Technology (Beverly, MA, U.S.A.). The [32P]dCTP and [γ-32P]ATP were obtained from NEN (Boston, MA, U.S.A.). Aldosterone, curcumin and spironolactone were purchased from Sigma-Aldrich (Deisenhofen, Germany). PD98059 was purchased from Calbiochem (Darmstadt, Germany). SP600125 was purchased from Biomol Research Laboratories (Plymouth Meeting, PA, U.S.A.). All supplements for cell culture were purchased from Life Technologies (Gaithersburg, MD, U.S.A.).
Cell culture
Rat mesangial cells were isolated from glomeruli of normal renal cortex as described previously (14). Mesangial cells were grown in RPMI1640 medium supplemented with 10% heat-inactivated FBS, 10 mM HEPES, and an antibiotic/antifungal solution.
TGF-β1 ELISA
After a 24 hr starvation, confluent cells were stimulated with aldosterone (0.2 ng/mL, 1 ng/mL, 5 ng/mL) for 24 hr. The conditioned medium was collected, centrifuged at 10,000 xg (4℃) for 3 min and stored at -70℃ until assayed. For the time course, confluent cells incubated in the serum free medium were incubated with or without aldosterone (5 ng/mL) for the indicated time points. For TGF-β1 ELISA, the conditioned medium was incubated with 1N HCl for 60 min to activate latent TGF-β1, and then neutralized with 1N NaOH. Anti-TGF-β1 Ab (1 µg/mL) and biotinylated Anti-TGF-β1 Ab (300 ng/mL) were used. The TMB microwell peroxidase substrate system (Kirkegaard & Perry Laboratories, Gaithersburg, MD, U.S.A.) was used as an enzyme substrate and the reaction was followed by measurement of OD450.
Isolation of RNA and TGF-β1 Northern blot analysis
Serum-starved confluent cells were stimulated with aldosterone (5 ng/mL) for the indicated time points. RNA was isolated by one-step guanidium-thiocyanatephenol-chloroform extraction. RNA quantification was determined by absorbance at 260 nm. Total RNA was fractionated by electrophoresis on a 1% agarose-formaldehyde gel, blotted by capillary transfer on a nylon membrane (Gene Screen; NEN, Boston, MA, U.S.A.), and cross-linked by UV irradiation (UV Strata-linker 1800; Stratagene, La Jolla, CA, U.S.A.). The membrane was probed with rat TGF-β1 cDNA labeled by random priming using a commercial kit (Amersham, Buckinghamshire, U.K.) and [32P]dCTP. The blot was prehybridized at 42℃ for 2 hr in 50% formamide, 0.1% SDS, 2×Denhardt's solution, 5×standard SSPE, and 0.1 mg/mL salmon sperm DNA. Probe (106 cpm/mL) was added to the prehybridization solution, and the blot was hybridized overnight at 42℃. The blot was then washed twice for 10 min each at 55℃ in 2×standard saline citrate and 0.1% SDS, and autoradiography was performed with Fuji radiograhpy film (Tokyo, Japan) and an intensifying screen at -70℃. The rat TGF-β1 probe was then removed by boiling, and the same blot was rehybridized to a cDNA probe encoding for the GAPDH. The intensity of the blots was quantified by densitometric analysis.
Electrophoretic mobility shift assay (EMSA)
Confluent cells were starved for 24 hr and then were stimulated with aldosterone (5 ng/mL) for 15 min. Nuclear extracts were prepared from the treated cells that were pelleted (-500 xg, 10 min, 4℃) and resuspended in hypotonic buffer (10 mM HEPES, pH 7.9, 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT, 0.5 mM PMSF, complete protease inhibit mixture tablets) and incubated for 10 min on ice. Cells were lyzed by the addition of Nonidet P-40 (final concentration of 0.1%). The nuclear pellet was resuspended in extraction buffer (20 mM HEPES, pH 7.9, 0.4 mM KCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, complete protease inhibit mixture tablets) and incubated for 10 min on ice, and the tubes were centrifuged at 14,000 xg (4℃) for 10 min. The supernatants were stored at -70℃ until used. The protein content in nuclear extracts was measured by the Bradford method (Bio-Rad, Hercules, CA, U.S.A.). The oligonucleotide containing the AP-1 consensus binding sequence was radiolabeled with [γ-32P]ATP by T4 polynucleotide kinase (Roche, Mannheim, Germany) for 30 min at room temperature. Unincorporated label was removed with a Chroma spin STE-10 column (Clontech Laboratories, Palo Alto, CA, U.S.A.). The binding reaction was performed for 30 min at room temperature in a total volume of 20 µL containing binding buffer (50 mM Tris, pH 7.5, 250 mM NaCl, 2.5 mM EDTA, 20% glycerol, 2.5 mM DTT, and 5 mM MgCl2), 5 µg of nuclear protein extracts, 1 µL of 32P-labeled oligonucleotide, and 50 µg/mL poly (dI-dC) (Roche). The DNA-protein complexes were fractionated by electrophoresis over 6% polyacrylamide gels in 1x Tris-boric acid-EDTA buffer. The gels were dried and exposed to radiograhy film for autoradiography.
Western blot analysis
Confluent cells were starved for 24 hr and then were stimulated with aldosterone (5 ng/mL) for 10 min. The treated cells were lyzed for 10 min on ice in lysis buffer (50 mM Tris, pH 7.5, 40 mM NaCl, 1% Triton X-100, 2 mM EDTA, 1 µg/mL leupeptin, 2 mM DTT, and 1 mM PMSF). Lysates were cleared by centrifugation at 14,000 xg (4℃) for 10 min. Total protein was quantified by the Bradford method. Equal amounts of lysates were fractionated by 10% SDS-PAGE and electrotransferred to Bio-Blot nitrocellulose membranes (Bio-Rad). The membranes were blocked with TBS (pH 7.6)/5% nonfat dry milk/0.05% Tween 20, and were blotted with the indicated antibodies at 4℃ overnight. The membranes were incubated with 1/1,000 diluted HRP-conjugated secondary antibody (Amersham) at room temperature for 1 hr and visualized by an ECL kit (Amersham). The membranes were stripped in solution (62.5 mM Tris-HCl, pH 7.5, 20 mM DTT, and 1% SDS) for 5 min at 50℃ with agitation and reprobed with anti-total JNK or anti-total ERK1/2 antibody.
Statistical analysis
Data were expressed as the mean±SEM. Statistical comparisons between multiple groups were performed by ANOVA and Bonferroni's method was applied to control for multiple testing. Statistical comparisons between multiple time points were performed by repeated measure ANOVA, with a minimum value of p<0.05 considered to represent statistical significance.
RESULTS
Aldosterone Stimulates TGF-β1 Production
TGF-β1 protein was measured in the supernatants of rat mesangial cells stimulated with aldosterone (5 ng/mL) by ELISA. TGF-β1 was constitutively produced by rat mesangial cells and was stimulated by aldosterone, which was statistically significant at 24 hr and 48 hr (n=5, p<0.05; Fig. 1A). We then measured TGF-β1 protein in the supernatants of cells incubated with various concentration of aldosterone with or without angiotensin II (10-7 M) for 24 hr. Aldosterone stimulated TGF-β1 production in a dose-dependent manner. Angiotensin II also stimulated TGF-β1 production as previously described in the same cells (15). Furthermore, a combination treatment of the cells with aldosterone and angiotensin II additively stimulated TGF-β1 production significantly more than single treatment (n=5, p<0.05; Fig. 1B). To determine if the mineralocorticoid receptor mediates the aldosterone-induced TGF-β1 production, cells were pre-incubated with or without spironolactone (10-9 M) for 60 min and then stimulated with aldosterone (5 ng/mL) for 24 hr. Spironolactone abolished aldosterone-induced TGF-β1 production (n=5, p<0.05, Fig. 1C).
We also examined the TGF-β1 protein expression in the lysates of cells, which were stimulated with aldosterone for 24 hr by western blot analysis. Aldosterone significantly stimulated TGF-β1 protein expression in a dose-dependent manner, which was blocked by pre-incubation of cells with spironolactone (10-9 M) (n=5, p<0.01, Fig. 1D).
Aldosterone stimulates TGF-β1 mRNA expression
We measured TGF-β1 mRNA in the cells incubated with various concentration of aldosterone with or without angiotensin II (10-7 M) for 16 hr. Aldosterone significantly stimulated TGF-β1 mRNA expression in a dose-dependent manner. A combination treatment of the cells with aldosterone and angiotensin II additively stimulated TGF-β1 mRNA expression significantly more than single treatment (n=5, p<0.05, Fig. 2A). When cells were pre-incubated with spironolactone (10-9 M) for 60 min and then stimulated with aldosterone (5 ng/mL) for 16 hr, aldosterone-induced TGF-β1 mRNA expression was abolished (n=5, p<0.05, Fig. 2B).
Role for AP-1 activation in the aldosterone-induced TGF-β1 expression
The 5'-flanking sequences of rat TGF-β1 gene contains AP-1 binding site (16), suggesting a potential role for AP-1 in the regulation of TGF-β1 expression. However, it has not been clearly determined whether aldosterone stimulates AP-1 activity and whether aldosterone-induced AP-1 activity triggers TGF-β1 expression in mesangial cells. Therefore, we examined AP-1 activity in rat mesangial cells stimulated with aldosterone by EMSA using AP-1 consensus oligonucleotides. AP-1 DNA binding activity was significantly increased by aldosterone (5 ng/mL), which was maximal at 15 min (n=5, p<0.05, Fig. 3A). Supershift analysis indicated that the AP-1 complexes induced by aldosterone contained c-Jun and c-Fos proteins (Fig. 3B). The specificity of the AP-1 binding activity was tested using both an unlabelled consensus AP-1 oligonucleotiedes (cold probe) and a mutant AP-1 oligonucleotides, which has two base changes within the AP-1 binding motif (CA to TG), at the same molar quantity (50-fold excess) for competition. The specific band induced by aldosterone was deleted by preincubation of nuclear extracts with excessive amounts of unlabelled AP-1 oligonucleotides, but not by preincubation of nuclear extracts with mutant AP-1 oligonucleotides (data not shown). When the cells were preincubated with c-Jun/AP-1 inhibitor, curcumin (12.5 µM), for 15 min, the aldosterone-induced AP-1 activity was completely abolished (n=3, p<0.05, Fig. 3B). To examine whether aldosterone-induced activation of AP-1 is responsible for the increased TGF-β1 production, cells were pre-incubated with curcumin (12.5 µM) for 15 min and then were stimulated with aldosterone (5 ng/mL) for 24 hr and TGF-β1 was measured in the cell supernatants by ELISA. Curcumin completely inhibited the aldosterone-induced TGF-β1 production (n=5, p<0.05, Fig. 3C).
Role for JNK pathway in the aldosterone-induced TGF-β1 expression
The activation of JNK has been shown to be linked to AP-1 activity particularly to the phosphorylation of c-Jun (17). We examined the phosphorylation of JNK in the cells stimulated with aldosterone by western blot analysis. Aldosterone (5 ng/mL) phosphorylated both 46 and 54 kDa JNKs in mesangial cells from 2 to 10 min and the peak phosphorylation was seen at 10 min (n=5, p<0.05, Fig. 4A). When the cells were pre-incubated with spironolactone (10-9 M) and then stimulated with aldosterone, aldosterone-induced JNK activity was significantly inhibited (n=5, p<0.05, Fig. 4B). When the cells were pre-incubated with JNK inhibitor, SP600125 (20 µM), and then stimulated with aldosterone (5 ng/mL), aldosterone-induced TGF-β1 protein production was significantly inhibited (n=5, p<0.05, Fig. 4C).
Role for ERK1/2 pathway in the aldosterone-induced TGF-β1 expression
We examined the phosphorylation of ERK1/2 in the cells stimulated with aldosterone by western blot analysis. Aldosterone (5 ng/mL) phosphorylated both 42 and 44 kDa ERK1/2 in mesangial cells from 2 to 60 min and the peak phosphorylation was seen at 10 min (n=3, p<0.05, Fig. 5A). When the cells were pre-incubated with spironolactone and then stimulated with aldosterone, aldosterone-induced ERK1/2 activity was significantly decreased (n=3, p<0.05, Fig. 5B). In addition, when the cells were pre-incubated with ERK1/2 inhibitor, PD98059 (50 µM), and then stimulated with aldosterone (5 ng/mL), aldosterone-induced TGF-β1 protein production was inhibited (n=5, p<0.05, Fig. 5C).
DISCUSSION
In the present study, we demonstrated that aldosterone stimulated TGF-β1 expression in rat mesangial cells, partly through the induction of ERK1/2 and JNK phosphorylation and AP-1 DNA binding activity. These results suggested a mechanism involved in the anti-fibrotic effect of spironolactone which was demonstrated in the animal models of various renal injury as well as human chronic kidney diseases.
In our study, aldosterone induced transcription factor, AP-1 DNA binding activity, which led to TGF-β1 up-regulation in mesangial cells. Recently, the role of AP-1 activation in the regulation of TGF-β1 expression in mesangial cells has been described in a few studies. A positive regulatory region in TGF-β1 5'-flanking sequence, which is responsible for the induction of TGF-β1, has an AP-1 binding site. Oxidized LDL has been shown to induce TGF-β1 expression through the AP-1 binding site (16). In a study done by Ahn et al. (18), transfection of mesangial cells with AP-1 decoy oligodeoxynucleotides strongly inhibited high glucose- and angiotensin II-induced TGF-β1 expression. In addition, administration of AP-1 decoy oligodeoxynucleotides into streptozotocin-induced diabetic rat kidney using the hemagglutinating virus of Japan-liposome method also abolished TGF-β1 and plasminogen activator inhibitor-1 expression.
c-Jun contains a docking site for the JNK, which phosphorylate serines 63 and 73, thereby enhancing its transcriptional potential and stability (19). Activation of JNK results in greatly enhanced c-Jun transcriptional activity and induction of the AP-1 target gene (19). Most cells express two JNK isoforms, 45 kDa and 54 kDa in size and termed JNK1 and JNK2, which are highly similar in their modes of regulation (20). In our study, aldosterone rapidly induced phosphorylation of JNK and spironolactone abolished aldosterone-induced phosphorylation of JNK. In addition, both JNK inhibitor, SP600125, and spironolactone suppressed aldosterone-induced TGF-β1 production, suggesting that blocking JNK activation contributed to the inhibition of c-jun/AP-1 activation, and finally TGF-β1 expression.
In our study, aldosterone rapidly stimulated ERK1/2 phosphorylation which led to subsequent increase in the TGF-β1 production. Aldosterone-induced ERK1/2 activation has been demonstrated in different in vitro and in vivo studies. Aldosterone-induced ERK1/2 activation has been shown to stimulate collagen gene expression and synthesis in renal fibroblasts (21) to induce mesangial cell proliferation (22), and to increase mytogenic response synergistically with angiotensin II in vascular smooth muscle cells (23). However, a direct relationship between ERK1/2 activation and TGF-β1 up-regulation in mesangial cells has not been clearly determined. In an animal study using aldosterone/salt-induced hypertensive rats, chronic administration of spironolactone inhibited ERK1/2 activity and suppressed expression of TGF-β1, type I and III collagen, and monocyte chemoattractant protein-1 mRNA, which resulted in the amelioration of perivascular fibrosis and coronary microvascular hyperplasia (24). However, a direct relationship between ERK1/2 activation and TGF-β1 up-regulation was not confirmed in that study. Recently, the role for ERK1/2 in the stimulation of TGF-β1 expression was demonstrated in two different cells. In a study done by Huang et al. (25), renin induced rapid activation of cellular ERK1/2 in rat mesangial cells and an ERK kinase inhibitor significantly reduced the renin-induced ERK1/2 phosphorylation and the subsequent increase in TGF-β1 mRNA expression. Otsuka et al. (26) has reported that liposomes composed of phosphatidylserine activated ERK which led to the expression of TGF-β1 in macrophages.
In summary, our data provide an evidence that aldosterone stimulates TGF-β1 expression in rat mesangial cells partly by enhancing ERK1/2 and JNK activities and subsequent AP-1 activity. This study also provides an understanding of a part of intracellular mechanisms for proliferative and fibrotic effect of aldosterone in the inflammatory renal diseases.

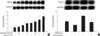



 XML Download
XML Download