

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Propafenone (2'-[3'(propylamino)-2-(hydroxy)propoxy]-3-Phenylptopiophenone hydrochloride) is a widely used antiarrhythamic drug. Electrophysiological studies in animal models have shown that propafenone reduces the maximum rate of rise and the amplitude of the action potential (1), and increases the duration of the action potential (2, 3). Furthermore, the drug has been shown to depress the transient outward current (Ito) in atrial myocytes of the rabbit and ventricular myocytes of the rat (4), the hyperpolarization-activated inward current (If) in isolated human atrial myocytes (5) and the delayed rectifier current (Ikr) in sinoatrial node cells and atrial myocytes of rabbits and ventricular myocytes of guinea pigs (3, 4, 6).
Voltage-operated potassium currents play important roles in shaping and terminating cardic action potentials. Although several potassium current components have been isolated, two types of currents can be distinguished: fast activating and inactivating currents, referred to as Ito, and delayed, more slowly inactivating currents, referred to as Ikr (7). For Ito, in particular, kv1.4 potasium channel has been identified or discussed, since the kv1.4 channel carries the Ito current which is a major contributor to the repolarizing currents terminating the cardic action potential. Several experiments have showed that the expression of kv1.4 channels in heart is altered under pathologic conditions associated with arrhythmias. Thus, 1) hyperthyroidism drastically decreased the protein level of kv1.4 in cultured newborn rat ventricular myocytes (8); 2) rats with infarcted myocardium showed reduced mRNA and protein levels of kv1.4 in different regions of left ventricular myocardium (9); 3) cardic hypertrophy in rats (induced by phorbol esters) increased the density of kv1.4 (10); 4) diabetic heart (induced by streptozotocin) yielded a increase of kv1.4 mRNA density and protein level in rat ventricle (11).
Therefore, understanding the molecular basis of the kinetic behavior of kv1.4 channels in response to changes in PH and [K]0 may be of considerable physiological important. We used an N-terminal deletion construct of kv1.4 (Kv1.4ΔN), which lacks rapid N-type inactivatin, but exhibits robust C-type inactivation (12, 13). In the present study, we aimed to investigate effects and mechanism of action of propafenone on the Kv1.4ΔN channel, and to examine the response to changes in PH and [K+]0.
MATERIALS AND METHODS
Molecular biology
The constructs and sequences of the cDNAs ferret Kv1.4ΔN (fKv1.4ΔN) used in this study have been previously described (12, 13). Ferret Kv1.4ΔN channels possess slow (C-type) inactivation (12, 13). Removal of residues 2-146 from the N-terminal domain (fKv1.4ΔN) results in the loss of the fast component of inactivation but leaves C-type inactivation.
Oocytes were collected from mature female Xenopus laevis under anaesthesia (immersion in 1.5 g l-1 tricaine). The follicular layer was removed enzymatically by placing the lobes in a collagenase-containing, Ca2+-free OR2 solution (mM: 82.5 NaCl, 2 KCl, 1 MgCl2 and 5 Hepes, pH 7.4, with 1-2 mg mL-1 collagenase (Type I, Sigma). The oocytes were gently shaken for about 2 hr and collagenase activity was then halted by bovine albumin as previously described (12). Defolliculated oocytes (stage V-VI) were then injected with transcribed cRNAs (up to 27-34 nL) and incubated at 18℃ for 24-72 hr in antibiotic-containing Barth's solution (mM: 88 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 MgSO4, 1.5 CaCl2 and 5 Hepes, pH 7.4) which was supplementd with penicillin (100 IU/mL).
Electrophysiology
Oocytes were clamped using a two-microelectrode bath clamp amplifier (CA-1B, Dagan Corp, Minneapolis, MN, U.S.A.). Microelectrodes were made from borosilicate glass tubing and had a resistance of 0.5-1 MΩ for the current electrodes and 1-2 MΩ for the potential electrodes when filled with 3 M/L KCl. During recording, oocytes were continuously perfused with control solution (mM: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2 and 10 Hepes, adjusted to pH 7.4 with NaOH) or high extracellular potassium concentration solution (mM: 98 KCl, 1 MgCl2, 1.8 CaCl2 and 10 Hepes, adjusted to pH 7.4 with NaOH) or normal extracellular potassium concentration solution (mM: 93 NaCl, 5 KCl, 1 MgCl2, 1.8 CaCl2 and 10 Hepes, adjusted to pH 7.4 with NaOH) or low extracellular potassium concentration solution (mM: 97 NaCl, 1 KCl, 1 MgCl2, 1.8 CaCl2 and 10 Hepes, adjusted to pH 7.4 with NaOH) The PH value of this solution was changed in some experiments and was set 6.0 with HCL and to 8.0 with NaOH. Whenever propafenone was used, 10 min of perfusion time was used to allow equilibration of propafenone with the oocyte. Propafenone (as hydrochloride; Sigama, isenhofen, Bayern, Germany) in final concentrations of 0.01 µM/L up to 1,000 µM/L was added to the bath solution. Solutions were applied with a concentration-clamp techinique for oocytes (14). All experimnts were performed at days 1 and 3 after injection of cRNA and were carried out at room temperature (22±1℃). Currents were filtered at 0.5 kHz.
Data analysis
Data were recorded on videotape using an A/D VCR adaptor (Axon Instruments, Union City, CA, U.S.A.) and were digitised and analysed directly using pCLAMP 9 software (Axon Instruments, Union City, CA, U.S.A.), Microsoft Excel (Microsoft, Redmond, WA, U.S.A.). Unless otherwise stated, raw data traces from two-microelectrode voltage-clamp recordings were not leakage or capacitance subtracted.
The potassium current amplitude was measured at the peak of current obtained during the depolarizing voltage step. Conductance-voltage relations were obtained by normalizing the conductance data to the maximal value under control conditions and by fitting the data to the Boltzmann equation y=Gmax (1+exp [(V1/2-V)/b]), where y is the normalized conductance, Gmax is the normalized maximal conductance, V1/2 is the potential of the half-maximal conductance, V is the voltage, and b is the slope factor. Concentration-response curves were determined by fitting the mean current values at different PROP concentrations to the Langmuir equation y=(KD/c)nH/[1+(KD/c)nH], where y is the fraction of control current, KD is the half blocking concentration, c is the concentration of PROP, and nH is the Hill coefficient. The voltage dependence of block was determined using the KD values that were obtained for these calculations from the fractional current (f), measured as the current in the presence of propafenone (PROP) at a concentration [D] of 100 µM and under control conditions (ICTRL) at the end of the voltage step: f=IPROP/ICTRL and KD=[D]×f/(1-f). Data are shown as means±S.E.M. Confidence levels were calculated using Student's paired t-test.
RESULTS
With application of depolarizing voltage steps to oocytes injected with fKv1.4ΔN cRNA, outward currents appeared at potentials of around -40 mv (Fig. 1A). The currents during the voltage step increased slowly. The currents reached the maximum at around +50 mv (Fig. 1B). The administration of 100 µM/L propafenone resulted in a decreased of the sustained currents (Fig. 1B, open squares).
The block of the sustained currents showed a voltage dependence with the largest blocking effect at the highest amplitudes (Fig. 2). The kinetics of the fKv1.4ΔN currents were affected by propafenone depending on voltage. The currents were reduced by propafenone over the whole potential range (Fig. 2, n=10). At a potential +50 mv, 100 µM/L propafenone reduced the amplitude of the current to 41% of the control value. The block appeared to be voltage-dependent and to be increased in the positive potential range (from 61±4% fraction of control current amplitude at -40 mv to 32±2% at +50 mv with 100 µM/L propafenone). Voltage dependence of propafenone-induced block of fKv1.4ΔN currents was presented after application of 10 µM/L, 100 µM/L, and 1,000 µM/L propafenone, respectively. At a potential +50 mv, 1,000 µM/L propafenone (Fig. 2, triangles) reduced the amplitude of the current more than that of 100 µM/L (Fig. 2, squares) and 10 µM/L (Fig. 2, diamonds).
Frequent-dependent block of fKv1.4ΔN channels induced by propafenone can be identified in Fig. 3. The current induced by the first pulse of the pulse train in the presence of 100 µM propafenone was dramatically decreased comparing with that in the pre-drug control (p<0.05, n=5) and then quickly reaches a steady-state current level, when the cell was continuously stimulated at 1 Hz. All these behaviors reflected the process of 100 µM propafenone blocking fKv1.4ΔN (at +50 mV) and showed a use-dependent kinectics. It showed with the increasing of numbers of opening channels, the block of propafenone on fKv1.4ΔN increased, that is open channel block.
Fig. 4 showed the effect of propafenone on fKv1.4ΔN recovery from inactivation with a standard two-pulse protocol. The fKv1.4N channel in propafenone recovered more slowly than the channel in control (Fig. 4A) (p<0.05, n=5). With the increasing of inter-pulse duration, the normalized current increased without decreasing at the end of the curve. It can be concluded that closed state inactivation does not exist and just open channel block exists. The 50% recovery time was 980±89.39 ms for controland it was 1,653.15±108.45 ms for drug-treated group (Fig. 4B) (p<0.05, n=5).
This change in time constant with propafenone suggests that prpafenone can accelerate the inactivation of fKv1.4ΔN currents (Fig. 5, n=8). At a potential of +50 mv the time constants of monoexponential fits were found to 4,494±508 ms under control conditions and 704±179 ms with 100 µM/L propafenone. There was significant difference between the time of constants for fKv1.4ΔN under control conditions and propafenone with 100 µM/L.
The concentration dependence of the propafenone effect was obtained by eliciting fKv1.4ΔN currents at propafenone concentrations between 1 µM/L and 1,000 µM/L and by fitting the amplitudes of the currents with Langmuir equations (Fig. 6A, n=5). The maximal block of the fKv1.4ΔN currents was 93% and was almost completely blockade by 1,000 µM/L. The concentration of half-maximal block (IC50) was 121 nM/L for the fKv1.4ΔN by propafenone.
The concentration dependence of the propafenone effect was also studied in high-potassium solution (98 mM), normal-potassium solution (5 mM) and low-potassium solution (1 mM) in Fig. 6B (n=5). Under control conditions of different extracellular potassium concentration, the fractioins of inhibition of propafenone at a concentration of 1,000 µM/L were 83%, 89%, and 94%, respectively. The Ic50 for propafenone block of the fKv1.4ΔN currents in 98[K+]0, 5 mM[K+]0, and 1 mM[K+]0 were 153 µM/L, 128 µM/L, and 94 µM/L, respectively. The Ic50 values were changed slightly and have no significant statistically differences.
Effects of extracellular PH on the concentration dependence of the propafenone effect were studied on fKv1.4ΔN currents in extracellular potassium concentration of 98 mM[K+]0, 5 mM[K+]0, and 1 mM[K+]0 (Fig. 7, n=5). With extracellular potassium acide solution (PH 6, triangles), neutral solution (PH 7.4, squares) and alkaline solution (PH 8, diamonds), propafenone was applied in concentration between 10 µM/L and 1,000 µM/L and the currents were fitted with Langmuir equations.
As described in literature, the kinetics of the fKv1.4ΔN currents were PH-dependent and the deactivation was accelerated with acidification and slowed down with alkalization in high-, normal- and low- extracellur potassium concentration. The sensitivity of the fKv1.4ΔN to propafenone, however, was altered with PH. Whereas the IC50 value increased in acidic solution to 463 µM/L, it was decreased in the alkaline extracellular solution to 58 µM/L (differences statistically significant) for the high extracellular potassium concentration.
DISCUSSION
Kv1.4 channel mediate a slowly recovering outward current with upregulated expression during ischemia. Therefore, understanding the molecular basis of the kinetic behavior of kv1.4 channel in response to changes in PH and [K+]0 may be of considerable physiological importance.
Ion channels undergo substantial physical rearrangements during gating when they transition between conducting and non-conducting states. This rearrangements can have a strong influence on channel affinity and drug binding, access to binding sites, and the environment in which they are in, can be altered considerably.
The result of study allow to infer on the mechanisms of action of propafenone at the fKv1.4ΔN channel molecule. The voltage dependence of the block suggests that the amount of block increased with the relative conductance and thus with the open probability of the fKv1.4ΔN channel. This means that the block because more effective when the period of time was prolonged in which fKv1.4ΔN channel was in the open state. The finding allow the conclusion that propafenone blocks the fKv1.4ΔN channel in the open state. This is consistent with blocking mechanisms of propafenone found for kv1.5 (15) and HERG channel (16). The use dependence of the block shows with the increasing of numbers of opening channels, the block of propafenone on fKv1.4N increased, that is open channel block.
Previous studies have shown that propafenone binds to the open conformation of HERG channels, and that propafenone inhibit the transient outward K current in human atrium, but this study is the first to examine the relationship between propafenone binding and C-type inactivation in a voltage-gated K channel.
Concerning the site of action of propafenone on fKv1.4ΔN channel, the results are not decisive. However, there is one line of evidence might be interpreted to point to a block by propafenone from the intracellular with lowering of the PH value. In acidic solution the protonation of propafenone is increased. Since for an action from the inside, propafenone has to cross the cell membrane and since it is more likely that unchanged molecules can transverse into the cell interior, a changing of propafenone by protonation can be assumed to reduce the concentration of propafenone within the cell, thus causing smaller blocking effects. For this interpretation, however, it has to be taken into consideration that the change of efficacy of propafenone can be assumed to be affected by the time the channel remained in the open state by reducing the probability of open channel block. Almost the same result has been found for the effect of the antiarrhythmic drug quinidine at the Kv1.5 channel and has been taken as an argument for an action from the inside.
There are two main hypotheses concerning the link between occlusion of the intracellular pore by drug or lipophilic peptides and the coupling to C-type inactivation: the "permeation" mechanism and the "allosteric" mechanism. The permeation mechanism (17) is based on the idea that blocking permeation directly effects C-type inactivation. The reduction in [K+]0 means fewer K+ ions bind to the extracellular binding site, so there is no hindrance to the channel adopting the C-type inactivated state (17). The allosteric mechanism (18, 19) proposes that the effect of reducing the K+ accumulation is negligible compared to the contribution from a direct physical interaction between events at the intracellular pore and C-type inactivation, which are mediated by the channel protein. C-type inactivation involves a conformational change at the extracellular mouth of pore (20). However, considerable evidence is emerging to indicate that C-type inactivation also involves major conformational changes on the intracellular side of the channel. The channel was regarded as an integrated unit in conformational terms, i.e. a conformational change at a remote site may result in allosteric changes throughout the protein. This leads to the results that an action at the intracellular face of the channel can affect an extracellular event such as C-type inactivation.
Transmembrane communication with allosteric mechanisms connecting physically remote sites has already been demonstrated for KV1.4 channels, eg. Mutating a valine at the C-terminal end of S6 has a dramatic effect on C-type inactivation of the KV1.4 channel (21). This kind of transmembrane communication is difficult to reconcile with the permeation mechanism.
Elevation of [K+]0 can reduce the affinity for drug binding by a direct (electrostatic or knock-off) effect or by an indirect effect via modification of the pore region by some kind of conformational change (allosteric effect), or by a combination of both (22). If the direct electrostatic effect was the basis of the interaction, elevating [K+]0 should reduce propafenone binding to Kv1.4ΔN channel. However, our data show that increasing [K+]0 had no significant effect in IC50 of propafenone for fKv1.4ΔN. This finding cannot be reconciled with the permeation hypothesis. This suggests that the modulation of drug binding by [K+] is not the result of an electrostatic interaction. The allosteric mechanisms dominate coupling in fKv1.4ΔN channels, and, by extension, may dominate in the wild type channel.
Recent studies have shown that propafenone blocks the kv2.1 channel in the open state from the intracellular side by entering the inner vestibule of the channel. These results are consistent with a direct interaction of propafenone with the lower part of the pore helix and/or residues of S6. These data show there is a link between C-type inactivation mediated pore closure and the propafenone-binding sites in the intracellular vestibule region. Since the orientation of S6 is critical to drug binding (23), and recent studies have shown a link between this domain and C-type inactivation (21), This suggests that S6 moves during C-type inactivation. Therefore, the conformational change accompanying C-type inactivation includes more of the channel than previously thought and has allosteric effect on the intracellular propafenone-binding site. The molecular sites of action of propafenone on fKv1.4ΔN channel is not fully clear. Previous studies have shown that PH change influence of c-inactivation involves the change on H508 and K532, and that a model of kv1.4 pore based on the crystal structure of KcsA, shows that H508 and K532 lie close together. Our study concludes that propafenone acts from the intracellular side of the membrane, and may result in a structural change near the sites of H508 and K532. Several regions have been found that when replaced by the corresponding parts of the Kv1.2 channel, reduced the sensitivity of the PROP for Kv2.1 channel (24). Replacement of the intracellular termini and the intracellular linker S4-S5 induced moderate effects. Because there is growing evidence that these parts of the channel form an intracellular compartment placed near membrane-associated part of the channel pore (25), the intracellular termini and the S4-S5 linker might impair the access of PROP to its proper site of action in the internal mouth of the channel. A complete abolition of PROP sensitivity of Kv2.1 (to the level of Kv1.2) was found with exchanges of the linker S5-S6, as well as with the S6 segment itself. Based on radiography analysis of the KcsA potassium channel, these parts can be assumed to form the inner wall of the pore proper (26). The intracellular amino and carboxyl terminus and the intracellular linker S4-S5 reduced the blocking effect of propafenone, whereas the linker S5-S6, as well as the segment S6 of the Kv1.2 channel. In the linker S5-S6, this effect could be narrowed down to two groups of amino acids (groups 372 to 374 and 383 to 384), which also affected the sensitivity tetraethylammonium. In segment S6, several amino acids in the intracellularly directed part of the helix significantly reduced propafenone sensitivity (24).
Taking all the findings into consideration, we suggest that PROP enters the cell by allosteric mechanism across the cell membrane and acts from the intracellular side. PROR blocks the Kv1.4ΔN channel in the inner vestibule of the channel while it is in the open state. The access of PROP to its binding site can be impaired by intracellular side. Residues of segment S6 might contribute to this impairment or to the formation of the binding site, which most probably includes the lower part of the pore helix.
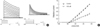





 XML Download
XML Download