

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Charcot-Marie-Tooth disease (CMT) is the most common inherited motor and sensory neuropathy, with an estimated prevalence of 1/2,500. CMT can be divided into one of two types: demyelinating (CMT1) or axonal (CMT2) (1). CMT1A duplication on chromosome 17p11.2-p12 is caused by an unequal crossover between two homologous repetitive elements that flank the 1.4-Mb region (2). Duplication is found in approximately 70% of patients with CMT1 (3). The peripheral myelin protein 22 (PMP22) gene is located within the duplication region, and alterations in the dosage of the PMP22 gene are the primary cause of the CMT1A phenotype (2). In contrast, hereditary neuropathy with liability to pressure palsies (HNPP) is caused by deletion of the same 1.4-Mb region, and patients with HNPP deletion have only one copy of PMP22 (4, 5). Myelin plays an important role in saltatory impulse transmission along neuronal extensions and communication between neurons and Schwann cells (6).
Schwannomas are tumors that originate in the Schwann cells of the peripheral nervous system, and the prevalence of schwannomas is estimated at 1/40,000 births (7, 8). Bilateral vestibular schwannomas and ophthalmological abnormalities are hallmarks of neurofibromatosis type 2 (NF2), whereas multiple fibromas are characteristic of neurofibromatosis type 1 (NF1) (8, 9). However, schwannomatosis characterized by multiple peripheral nerve and spinal schwannomas is clinically similar to NF1 and NF2, but has a distinct genetic background (9).
A patient with the rare combination of HNPP with PMP22 deletion and schwannomas has been previously reported (10). However, there has been no report of CMT1A in a patient with PMP22 duplication and schwannoma. In the present study, we present two patients in a CMT1A family (familial ID: FC270) with schwannomas of the spinal cord and median nerve.
CASE REPORT
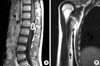
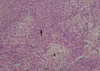
Patient 1: The proband (M/14 yr, Fig. 1: III-3) was admitted to our hospital due to radiating pain in his left leg lasting for two months. In addition, he had walking difficulty from 9 yr of age and had a steppage gait with foot dorsiflexion weakness, pes cavus, and hammer toes. Moreover, we found a palpable soft tissue mass with tenderness on the medial side of the right middle humerus. A neurological examination performed at the age of 14 yr revealed weakness of the distal muscles of the lower limbs. Vibration and pain perceptions were reduced in the distal upper and lower limbs, and stretch reflexes were decreased. Nerve conduction studies showed marked reduction in motor and sensory nerve conduction velocities (NCVs) consistent with demyelinating neuropathy. When stimulating at the elbow, right median motor NCV was 21.1 m/sec (normal ≥50.5 m/sec), and compound muscle action potential (CMAP) was 11.6 mV (normal ≥6.0 mV), and left motor NCV 19.8 m/sec, CMAP 9.1 mV. Right median sensory NCV between finger and wrist was 18.9 m/sec (normal ≥39.3 m/sec), and sensory nerve action potential (SNAP) was 8.9 µV (normal ≥8.8 µV), and left sensory NCV 18.2 m/sec, SNAP 7.4 µV. During stimulation at the axillary level, right median motor NCV was 21.1 m/sec (normal ≥51.2 m/sec), and CMAP was 9.0 mV (normal ≥6.0 mV), and left motor NCV 23.0 m/sec, CMAP 9.5 mV. Right sensory NCV between the elbow and axilla was 20.9 m/sec (normal ≥48.0 m/sec), and SNAP was 13.0 µV (normal ≥13.0 µV), and left sensory NCV 21.6 m/sec, SNAP 4.5 µV. Magnetic resonance imaging (MRI) scans of the thoracolumbar spine revealed a contrast-enhancing intradural extramedullary mass (1.8×2.4 cm) at the T12-L1 level (Fig. 2A). Surgery was performed, and the spherical mass was removed. The radiating pain in his leg had completely resolved one month after surgery. The histopathological features of this mass included crowded spindle cells with palisading nuclei and loosely arranged spindle cells in abundant myxomatous matrix areas (Fig. 3). We also found another schwannoma (1.5×2.0 cm) adjacent to the right median nerve at the level of the middle humerus (Fig. 2B).
Patient 2: The mother of the proband (F/40 yr, Fig. 1: II-6) complained of walking difficulty. She began to experience weakness in both lower extremities at 20 yr of age. At 25 yr, she had numbness and pain in her right hand and underwent surgery for carpal tunnel syndrome, which revealed the presence of a schwannoma. The pain decreased after surgery, and her symptoms had resolved completely three months later. A neurologic examination performed at the age of 40 yr revealed moderate weakness and atrophy in the distal muscles of the lower limbs and pes cavus in both feet. All of her sensory modalities were impaired, and all muscle stretch reflexes were absent. When stimulating at the elbow, median motor NCV was 20.0 m/sec, and CMAP was 9.5 mV, but SNAPs were not recordable. In this FC270 family, we identified duplication of the PMP22 gene in nine family members by genotyping six microsatellites within the 17p11.2-p12 duplication region (11). The duplication was well cosegregated with familial members diagnosed with demyelinating CMT1 (Fig. 1). However, no causative mutation in the NF1 gene was identified not only in patients with CMT1A alone, but also in those with the both CMT1A and schwannoma.
As neurofibromatosis was considered, additional ophthalmological and dermatological examinations and imaging studies of the brain and spinal cord were performed in the proband (III-3), his sister (III-4), mother (II-6), and aunt (II-8). These tests showed normal findings without any evidence for NF type 1 or 2.
DISCUSSION
We present two patients in a family with an unusual combination of CMT1A with PMP22 duplication and schwannomas. The CMT1A duplication was found to have an autosomal dominant pattern of inheritance. Schwannomas were found in the proband and his mother, which indicated a genetic predisposition, acting in an autosomal dominant manner.
The characteristics of NF1 are cafe au lait patches, neurofibromas, Lisch nodules and bony dysplasia. The NF1 gene was identified on chromosome 17q11.2 (8); however, no causative mutation in NF1 was found in the proband. In addition, NF2 is a rare autosomal dominant neurocutaneous disease characterized by bilateral vestibular schwannomas (9). No further evidence for NF1 or NF2 was found in either of our patients. Multiple schwannomas in the absence of other NF2 stigmata are characteristic of schwannomatosis, a newly described syndrome with genetic aberrations on chromosome 22 outside of the NF2 locus (9).
Patient 2, who was initially diagnosed with carpal tunnel syndrome, had complained of numbness and pain in her hand, and we finally found a schwannoma. In addition, her son had radiating pain from the proximal thigh to the distal foot, and MRI and histology revealed a schwannoma. Considering the proband's family history, we carefully examined his entire body and found a mass on his right arm. Imaging of the humerus revealed a secondary schwannoma. Therefore, in nerve tumors, careful examination would be helpful for the detection of causative lesions, such as schwannomas.
Hechkmann et al. reported a HNPP patient with a PMP22 deletion and schwannoma, and they suggested the possibility that dysfunction of PMP22 may not only result in peripheral neuropathy but also promote the development of Schwann cell neoplasms (10). CMT1A and HNPP are peripheral neuropathies typically caused by abnormal dosages of PMP22. Interestingly, CMT1A as well as HNPP are associated with point mutations in PMP22 gene (12, 13). Although it seems that the occurrence of two diseases in the same patient (CMT1A or HNPP vs. schwannoma) might be merely coincidental, we could not completely ignore the possibility that the coincidence of two diseases might be due to a shared genetic background. Therefore, although further studies are needed to determine the association between schwannomas and the PMP22 gene, a common pathogenetic basis for the disturbance of the Schwann cell life cycle may be involved.

 XML Download
XML Download