

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Sinus histiocytosis with massive lymphadenopathy (SHML) also known as Rosai-Dorfman disease, is first reported in 1969 (1). SHML is usually seen in the first and second decade of life, with no specific gender, ethnic or socioeconomic predilection. Painless lymphadenopathy is the most frequent presenting symptoms and involved the cervical lymph node in up to 90% of patients. In approximately 40% of patients, extranodal lesions in the skin and soft tissues, upper respiratory tract, genitourinary tract, oral cavity, kidney, thyroid, breast, and bone can be found (2). Typically, the lymph node sinuses are expanded by a proliferation of histiocytes with abundant pale eosinophilic cytoplasm containing emperipolesis (3, 4). The S-100 stain is helpful in identifying the histiocytes of SHML (5). There is no specific treatment for the SHML. We report a case of a young man who was diagnosed with SHML involving cervical lymphadenopathy and pleural effusion.
CASE REPORT
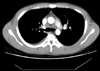
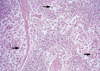
A 26-yr-old man presented with a firm, nontender palpable lymph node in his left cervical area. The patient had no specific past medical history. On admission his blood pressure was 120/80 mm Hg, pulse rate 80/min, respiratory rate 24/min, and body temperature 36.5℃. Routine laboratory investigations showed a normal complete blood count, serum chemical tests, and erythrocyte sedimentation rate (ESR). Serologic tests for cytomegalovirus, human immunodeficiency virus (HIV) and polymerase chain reaction (PCR) analysis of mycobacteria yield negative results. Serum immunoglobulin (Ig) levels were as follows: IgG, 1,680 mg/dL (normal 700-1,700 mg/dL); IgA, 117 mg/dL (normal 70-400 mg/dL); IgM, 129 mg/dL (normal 40-230 mg/dL); and IgE, 33.8 IU/mL (normal 0-100 IU/mL). Computed tomography of the chest showed mediastinal lymph nodes with calcification and pleural effusion (Fig. 1). Diagnostic thoracentesis was not performed because of scanty pleural effusion. Excisional biopsy of the cervical lymph node revealed dilated sinuses filled with histiocytes with abundant pale eosinophilic cytoplasm (Fig. 2). The histiocytes were positive immunoreactivities for CD68 and S-100 protein (Fig. 3). These findings were diagnostic of SHML. The patient had received prednisolone therapy (30 mg per day, PO). On follow-up the patient was well without symptoms and signs.
DISCUSSION
Sinus histiocytosis with massive lymphadenopathy is a rare disorder characterized by a nonmalignant proliferation of histiocyte within lymph node sinuses and lymphatics in extranodal sites. There is no evidence to support immunodeficiency, autoimmune disease or a neoplastic process for the etiology of the disorder. An association with Epstein-Barr virus (EBV), cytomegalovirus (CMV), Brucell, Klebsiella, or human herpes virus 6 has been suggested but not proven (6-8). Serologic tests for CMV, HIV, and PCR of mycobacteria were negative in our patient.
Most patients with SHML tend to have a chronic massive enlargement of cervical lymph nodes frequently accompanied by fever, elevated ESR, neutrophilia, polyclonal gammopathy (9). Our patient had no history of fever and laboratory investigations showed a normal complete blood count, ESR, immunoglobulin levels. Extranodal disease is seen in approximately 30% of patients. The most often affected extranodal sites include skin and soft tissues, upper respiratory tract, orbit, testicle, kidney, thyroid, small bowel, breast, and bone (2). Hepatosplenemegaly is uncommon.
Typical histopathologic findings of SHML are large polyclonal histiocytes with abundant pale, eosinophilic cytoplasm and display emperiopolesis. Mitoses are infrequent, although increased mitotic activity can be apparent occasionally (3, 4, 10).
SHML involving extranodal sites shows similar morphologic features to its nodal counterpart although more fibrosis and fewer histiocytes with emperiopolesis are encountered. The most useful immunohistologic marker for SHML histiocytes is the expression of the S-100 protein (11, 12). In addition, SHML histiocytes stain for CD68, CD64, alpha-1 antitrypsin, and interleukin-2 receptor and are negative for CD1a (5, 13, 14).
The clinical course of SHML is characterized by spontaneous resolution in most cases. There is no specific treatment for the SHML. Treatment is required when the condition is organ threatening or life threatening. For patients with high fever without other symptomatology steroid therapy may be instituted. Surgical debulking may be used in cases where vital organ function is compromised. The role of additional therapies, such as chemotherapy or radiation therapy, is minimal. However all patients deserve long-term follow-up, since the natural history of the disease is quite variable, usually alternating periods of exacerbations and resolutions or, rarely, pursuing a progressive course.

 XML Download
XML Download