

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Camurati-Engelmann disease (CED) (Mendelian Inheritance in Man 131300), also known as progressive diaphyseal dysplasia (PDD), is a rare disease that occurs in both genders with equal frequency and is inherited in an autosomal dominant pattern with variable penetrance and wide expressivity (1-3). Considerable variations in signs, symptoms, and severity between afflicted individuals exist within the same family (2, 3). The main clinical symptoms are pain in the legs, waddling gait, muscular weakness, and easy fatigability (1-3). Other associated symptoms include reduced muscle mass, exophthalmus, facial paralysis, hearing difficulty, and loss of vision. There are no specific laboratory findings or ultrastructural muscular abnormalities that are known but radiographic abnormalities are remarkably constant and distinctive. CED is a disease of endosteal and periosteal thickening and expansion of the diaphyses of the long bones. This results in cortical thickening, narrowing of the medullary cavity and a sclerotic and expanded diaphyseal segment (4, 5). Recently, the locus for CED was assigned to chromosomal region 19q13.1 and mutations have been found in the transforming growth factor-β1 (TGFB1) gene (1, 6). We report the first Korean CED family with 2 affected individuals confirmed by mutation analysis.
CASE REPORT
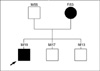
A 19-yr-old Korean male with a history of abnormal gait since the age of 2 was referred for molecular genetic testing. His symptoms included wide base waddling gait, proximal muscle weakness, pain in the extremities, and easy fatigability. He had no history of trauma, infection, or systemic illness. Physical examination showed no abnormalities in his reflexes and he was able to walk independently. The 53-yr-old mother of the proband had only mild limb pain with an onset after the fourth decade that subsided with steroid treatment but she had never previously received a conclusive diagnosis despite multiple visits to several hospitals. Other family members, including the proband's father and 2 younger brothers, did not show any symptoms of muscle weakness (Fig. 1).
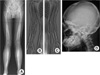
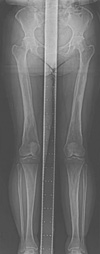
Radiographs of the proband showed cortical, periosteal, and endosteal bony thickening with narrowing of the medullary spaces confined to the diaphyses of the femur, tibia, and fibula (Fig. 2A). The epiphyses and metaphyses of the long bones are relatively spared. In the upper extremity, the ulna is rather severely affected than the radius (Fig. 2B, C). The humerus showed only mild cortical thickening. Some curvature changes were seen in both tibias with genu valgus changes of both knees. Scoliosis of the thoracic and lumbar spines with mild kyphosis of the thoracic spines was also observed. No other gross bony abnormality was seen in the skull (Fig. 2D). The chest radiography was unremarkable. Skeletal radiograph of the lower extremities in the proband's mother showed similar diaphyseal thickening and sclerosis of the femur and tibia with undermodeling of distal femur and proximal tibia (Fig. 3).
Laboratory findings in the patient indicated normal values for serum levels of electrolytes, calcium, and phosphate. Serum alkaline phosphatase was mildly elevated (139 IU/L; reference range, 42-117 IU/L) and peripheral blood analysis showed slight increase in ESR level (24 mm/hr; reference range, 0-15 mm/hr).
Muscle biopsy was performed at the patient's right vastus lateralis, which did not show any pathologic findings. Degenerating or regenerating fibers were not seen and immunohistochemical staining for dystrophin was normal along the membranes of the muscle fibers.
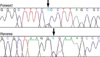
Genomic DNA samples after written informed consent were obtained from the patient and his mother. Exon 4 of the TGFB1 gene was targeted for molecular testing since this region is a mutation hot spot that includes approximately 80% of all mutations reported so far (1). Primers (forward: 5'-GGTTTGCTCCTTCCTTCCTC-3', reverse: 5'-GCAGATGGGAACACACACAC-3') were designed to amplify exon 4 of the TGFB1 gene and direct sequencing was carried out on an ABI 310 automated sequencer (Applied Biosystems, Foster City, CA, U.S.A.). Identical mutations of heterozygous G to A transition at cDNA position +653 (Fig. 4) were detected in both the proband and mother, which is predicted to result in an arginine to histidine substitution at amino acid 218 (R218H) near the carboxy terminus of the latency-associated peptide (LAP). This mutation had been previously reported in CED patients (7, 8).
DISCUSSION
Forty-five CED families have been described worldwide to date from Europe, Asia, Africa, North America, and Australia (1). CED patients usually have a positive family history showing an autosomal dominant mode of inheritance. Variable penetrance has given rise to a broad range of descriptions: lethal disease, disabled patients, and even patients whose radiographic abnormalities almost were incidental findings (2, 3). The age of presentation ranges from the first to the eighth decade depending on the clinical subtype (9). The extreme variability in phenotypical expression, both between families sharing the same mutation and among members of the same family, makes it difficult to detect possible genotype-phenotype correlations. Even in the present case, the patient and his mother showed different phenotypical expression despite having identical mutations in a region known to be associated with CED. Irrespective of the nature of the mutation, the age of onset and rate of disease progression appear highly unpredictable.
Prior to the definition of the molecular defect, Saraiva (10) proposed that CED might show anticipation because younger members were often more severely affected than their parents. Another report by Saraiva (11) supports the occurrence of anticipation in CED and widens the disease spectrum of this concept to bone dysplasias. The patient had a waddling gait since the age of 2 and was diagnosed in his second decade of life whereas the mother had only mild symptoms of late onset and was diagnosed during diagnostic workup of her son; therefore, genetic counseling may be recommended in successive generations in this family.
CED can be associated with neurologic symptoms, including headache, hearing loss, vertigo, and tinnitus, which have been previously noted in a study of a family through 4 generations with extensive skull involvement (2). The most common deficits are hearing loss, vision problems, and facial paralysis. In our study, the proband complained of recently developed facial muscle twitching. Despite having no abnormalities in radiologic findings of the skull, close monitoring for progression of symptoms should be considered in the patient.
The TGFB1 gene, which influences osteoblast and osteoclast function, is thought to act as a coupling factor between bone deposition and resorption (12). In addition, it has been reported that CED is caused by mutations in the gene encoding for TGFB1 (7, 8, 12, 13). With the presence of an activating mutation in the TGFB1 gene, the inhibiting effect of TGFB1 on osteoclast differentiation and activation and its stimulatory effect on osteoblast chemotaxis, proliferation, and differentiation will be enhanced (12). The missense mutation seen in both the patient and his mother (R218H) was located in a mutation hotspot, which represents 60% of the mutations (1). The majority of mutations detected in CED are missense mutations located in exon 4, coding for the region in the latency associated peptide surrounding the residues responsible for homodimerisation (Cys 223 and Cys 225), making up 82.2% of all mutations reported so far (1).
The incidence of CED tends to be underestimated since patients illustrate marked phenotypic variability. This patient had initially been tested for muscular dystrophy and the disease was ruled out with negative muscle biopsy findings. Like this case, many children who present symptoms of waddling gait, decreased muscle bulk and/or tone, fatigue, and weakness are often incorrectly diagnosed with muscular dystrophy or delayed diagnosis (2).
Diagnosis of CED is based on a combination of a thorough family history, physical examination, and radiologic studies with the utilization of molecular genetic testing for confirmation. Since TGFB1 is the only gene known to be associated with CED and the hot spot for mutations is known, diagnosis can be easily confirmed by mutation analysis. Moreover, with the identification of the first case of CED in a Korean family in addition to previously accumulated data of Asian cases, CED can presumably affect all races. Therefore, we need to keep a high index of suspicion in Korean patients with symptoms suggestive of CED.
In conclusion, this report describes the first confirmed Korean family with CED. Both the proband and his mother had symptoms and radiologic findings suggestive of CED and was confirmed with mutation analysis of the TGFB1 gene. The patient should be monitored closely for progression of the disease, including skull involvement.
 XML Download
XML Download