

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dystrophic epidermolysis bullosa (DEB) is caused by mutations in the COL7A1 gene encoding collagen VII, the major component of anchoring fibrils (1, 2). DEB is clinically characterized by mucocutaneous blistering in response to minor trauma, followed by scarring and nail dystrophy. It occurs as either an autosomal dominant (DDEB) or recessive (RDEB) trait, each form having unique clinical features and severity (3).
The most severe RDEB subtype, the Hallopeau-Simens (HS) type, shows a complete lack of expression of type VII collagen, whereas some collagen expression is found in the non-Hallopeau-Siemens (non-HS) type (4, 5). The most severe form, HS-RDEB, was usually caused by combinations of premature termination codon (PTC) mutations (6, 7). The non-HS type with the presence of some type VII collagen expression is generally due to a combination of PTC with mis-sense mutations including glycine substitution, in-frame deletion or a splice site mutation (8). These glycine substitution mutations cause various phenotypes. In the heterozygous state, some glycine substitution mutations cause a dominant DEB phenotype through a dominant negative interference or cause limited nail dystrophy or no phenotype at all (9-11). However, glycine substitution mutations lead to a moderately severe phenotype of RDEB when in the homozygous state or when combined with a PTC in the other COL7A1 allele (12).
In this study, we have systematically screened the COL7A1 gene, for mutational analysis in a RDEB Korean patient, 15-yr-old male, showing multiple erosion, crusts, milia formation on scar, mitten-like deformity of hand, and dysphagia, due to esophageal stricture, evidenced by esphagogram examination.
MATERIALS AND METHODS
Patient
A 15-yr-old male patient visited, suffering from multiple erosion, deformity of hand, and dysphagia. Several trauma-induced erosions and blister had developed mainly on his hands and trunk since infancy. His parents were non-consanguineous, and his family members including his siblings were clinically unaffected.
Primer design
For amplification of segments of genomic DNA, the primers were designed on the basis of flanking intronic sequences as previously reported by Christiano et al. (13). The oligomer primers were 20 nucleotides in length, contained no greater than three identical consecutive bases, minimal secondary structure, between 45 and 65% G/C content, and were free of the potential for primer-dimer formation. Amplimers ranged from 196 to 589 bp in size. Primers were synthesized using an automated oligonucleotide synthesizer (Bioneer Co., Daejeon, Korea).
Polymerase chain reaction (PCR) amplification of genomic DNA
For PCR amplification of genomic sequences, DNA was isolated from peripheral blood by the DNA extraction kit (Qiagen Co., Valencia, CA, U.S.A.), according to the manufacturer's recommendation. Purified DNA was used as a template in a reaction volume of 50 µL, containing 20 pM of each primer, 100 nM MgCl2, 20 mM of each nucleotide, and 2.5 U Taq polymerase (Perkin-Elmer, Cetus, MO, U.S.A.). The conditions for PCR were similar for each primer pair, except that the annealing temperature varied. After denaturation for 5 min at 94℃, each PCR cycle consisted of 94℃, 45 sec; annealing 55-61℃, 45 sec; 72℃ 45 sec; for 40 cycles. The PCR amplification products were examined on 2% agarose gels. The PCR amplimers were examined for detection of sequence variants by electrophoretic scanning techniques such as the heterodulpex analyses described below.
Heterodulpex analyses
Heteroduplex analyses were performed using conformation-sensitive gel electrophoresis (CSGE), as described by Ganguly et al. (14). For CSGE, 8 µL of each sample was denatured by heating at 98℃ for 5 min, followed by cooling at 68℃ for 1 hr. Loading dye (2 µL) was added and the samples were run on the CSGE gel overnight at 300 V in 0.25×TBE buffer (top chamber) and 1×TBE buffer (bottom chamber). Visualization of bands was then performed in ethidium bromide solution (1 ng/mL in 0.5×TBE).
Mutation detection
If bands with altered mobility were detected, the PCR products were sequenced. In some cases, the PCR products were subcloned into the TA-cloning vector according to the manufacturer's recommendation (Invitrogen, San Diego, CA, U.S.A.). Plasmid minipreparations were performed using the standard boiling methods, and several positive clones were sequenced by the dideoxynucleotide sequencing method (15). Alternatively, the PCR products were purified on a chromatographic column (QIAquick Spin column, Qiagen, Valencia, CA) and then subjected to direct dideoxynucleotide sequencing in an automated DNA sequencer (Applied Biosystems, Foster, CA, U.S.A.) with the same primers used for PCR amplification. The variant sequences in patients affected with DEB were compared with those in unaffected family members. Comparative sequence analysis with the normal COL7A1 template sequence, deposited in Gene Bank under accession No. L23982, was also performed.
RESULTS
Clinical features
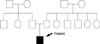
The patient showed the multiple erosion and crusts formation on trunk with scattered blister formation (Fig. 1A, B). He also was suffered from dysphagia due to stricture of gastroesophageal junction, observed by esophagography. His fingers and toes were shortened and contracted due to repeated blister formation, healing, and eventually scarring with mild fusion between webs (Fig. 1C, D). The pedigree showed that there is no affected person's among parents and relatives, except the patient (Fig. 2).
Mutational analysis of COL7A1
Heteroduplex analyses using amplified DNA from RDEB family revealed band shifts in the affected individual for PCR products spanning, within exon 116. The homoduplex band was seen in his mother. Unfortunately, we did not perform heteroduplex analysis of DNA from his father, due to the divorce state. Direct sequencing of the PCR products from the patient revealed G to T transition at position 8569. As a result, the codon for glutamic acid (GAG) is changed to a codon for stop (TAG), and this mutation was designated c.8569G>T and did not present in the maternal DNA samples. (Fig. 4A). Furthermore the mutation of c.8569G>T was confirmed by restriction enzyme (XspI), specifically recognizes and digests in CTAG nucleotide sequence. A further study revealed that G to A transition at position 4879 within exon 51. As a result, the codon for valine (VAL) is changed to a codon for isoleucine (Ile), and this mutation was detected both patient and mother DNA samples. Thus, our patient has a premature termination codon (Glu → Stop, 2857) mutation in one allele and missense mutation (Val → Ile, 1627) in the other allele. The mother and father may be carrier state and remain unaffected clinically (Fig. 5).
DISCUSSION
EB is a group of inherited skin diseases characterized by blistering and scarring of the skin after mild trauma. DEB is transmitted in an autosomal dominant or recessive pattern, both forms showing tissue separation beneath the basement membrane at the level of the anchoring fibrils (16). Type VII collagen molecules, major component of anchoring fibrils, are homotrimers, each pro α1 (VII) polypeptide chain containing a central triple-helical collagenous domain (145 kDa in size) flanked by both a large (145 kDa) amino-terminal noncollagenous (NC-1) domain and a small (30 kDa) carboxy-terminal noncollagenaous (NC-2) domain (17). The NC-1 domain consists of submodules with homology to an adhesive protein, including a matrix-protein motif, a fibronectin like domain, and a von Willebrand factor A domain (18). The NC-2 domain contains conserved cysteins involved in the formation of disulfide linkage between type VII collagen homotrimers (19, 20). In the conversion of procollagen VII to collagen VII, two procollagen VII molecules form an anti-parallel dimmer with eight cysteines in the carboxyl-terminal NC-2 domain, which is finally removed at the procollagen C-proteinase cleavage site. RDEB comprises several forms, the most severe of which is the Hallopeau-Siemens type (HS-RDEB). Other forms of RDEB comprise the mitis (M-RDEB), the inverse, and the localized types. It has been suggested that the nature and/or the location of mutations in COL7A1 determine the variations in clinical phenotype and type VII collagen expression. PTC mutations are silent when in a heterozygous state, but when homozygous or combined with another PTC mutation in other alleles, the consequences are severe in terms of skin fragility, as illustrated by HS-RDEB (9-12). In support of this interpretation, a majority of mutations reported in HS-RDEB result from PTC, which are thought to lead to null alleles through PTC-mediated mRNA decay (21). PTC association with either a glycine substitution, a slice-site mutation, or, a delayed termination codon have also been characterized in clinically moderate form of RDEB (12, 22, 23).
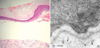
It has been known that Hallopeau-Siemens variant RDEB involves PTC mutations on both COL7A1 alleles leading to an absence of collagen VII and no detectable anchoring fibrils. However, in this study, electron microscopy examination provided evidence that the synthesis of rudimentary anchoring fibrils was still possible although its amount was considerably reduced. We hypothesized that some anchoring fibrils were formed in spite of the PTC mutations on COL7A1 allele, and maybe that is why the skin fragility and scar formation was not so severe in our patient. Actually, our patient showed a PTC (c.8569G>T) in exon 116. Exon 116 is located in the middle of the NC-2 domain, and we hypothesized that the truncated procollagen VII from the mutant allele with a PTC in exon 116 included two out of eight cysteines needed for disulfide bond formation, and hence a few functional anchoring fibrils could be formed. It is possible that the mutant allele of c.8569G>T may be inherited from father, due to the mother allele is normal. However, it still remains to be determined this mutant allele is really inherited from father. The both patient and mother have another novel mutation that G to A transition at position 4879 within exon 51; the codon for valine (VAL) is changed to a codon for isoleucine (Ile), and resulting in the substitution is occurred within the collagenous domain that form the collagen VII triple helix. In this study, the mother did not show any signs of mucocutaneous and nail involvement despite a careful reexamination. It still needs to investigate the maternal mutation c.4879G>A will be interpreted as a silent mutation or altered splicing.
In summary, in this study, we reported a moderately severe RDEB patient, who has the compound mutation of c.8569G>T in exon 116 and c.4879G>A within exon 51. We hope that this data contribute to the expanding database on COL7A1 mutations in DEB, and accumulating mutational data on patients with RDEB have been proved useful in providing more precise clinical information, particularly in defining diagnosis and in improving genetic counseling.



 XML Download
XML Download