

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Chronic granulomatous disease (CGD) is a rare hereditary disorder characterized by recurrent life-threatening bacterial and fungal infections (1). The underlying defect in CGD is an inability of phagocytes to produce reactive oxygen species (ROS) as a result of defects in NADPH oxidase (2). NADPH oxidase is composed of several structural and regulatory proteins. Two of these, gp91-phox and p22-phox, are integral membrane proteins that form flavocytochromeb558, the electron-transport center of the oxidase. The cytosolic proteins, p40-phox, p47-phox, and p67-phox, exist as a tight complex in the cytosol of resting phagocytes (3, 4). After ingestion of microbes into phagosomes, the cytosolic proteins translocate to the membrane to complex with the flavocytochrome to form activated NADPH oxidase, which generates ROS (5).
CGD is a genetically heterogeneous disease caused by defects in the genes encoding any of the NADPH oxidase components. Approximately 60% of CGD patients showed an X-linked inheritance, which is caused by mutations in the CYBB gene encoding gp91-phox. Autosomal recessive forms of CGD are caused by mutations in one of three genes: the CYBA (p22-phox) gene, the NCF1 (p47-phox) gene, and the NCF2 (p67-phox) gene. Mutations in these three genes collectively account for about 30-40% of CGD patients (6). Mutations in the p67-phox and p22-phox genes are less common, and to date no patients have been reported with defects in p40-phox (6).
Although the prevalence of CGD may actually be higher than reported because of the under-diagnosis of milder phenotypes, the estimated prevalence of CGD is between 1 in 200,000 and 1 in 250,000 individuals, with variable occurrence in different countries (6-11). According to a collation by the Korean College of Pediatric Clinical Immunology, the prevalence of CGD from 2001 to 2005 in Korea was 0.9 in 1,000,000 individuals. Most regions of Korea had similar prevalence of CGD (from 0.4 to 1.7). The prevalence of CGD on Jeju Island was a surprising 20.7, showing 10-50 times higher than that in other regions of Korea (paper in preparation). The aim of this study was to examine the genotype of CGD patients on Jeju Island.
MATERIALS AND METHODS
Patients
Between January 1993 and December 2007, the available data for CGD patients on Jeju Island were obtained from the medical records of Jeju National University Hospital. The diagnosis of CGD was based on a history of recurrent infections and previously affected family members. The diagnosis was confirmed by an impaired phorbol myristate acetate (PMA)-stimulated nitroblue tetrazolium (NBT) test and/or dihydrorhodamine-1,2,3 (DHR) flow cytometry assay. Twelve patients with CGD (3 males and 9 females) from 10 unrelated families were enrolled for Western blot and mutation analysis. All available family members were analyzed for confirming the heterozygous status.
This study was approved by the Institutional Review Board of Jeju National University Hospital (2006-14), and informed consent for genetic analysis was obtained from all patients and/or their parents.
DHR flow cytometry
DHR flow cytometry was used to evaluate the generation of ROS by phagocytic cells as previously described (12). Red blood cells were lysed by mixing 400 µL of whole blood with 4 mL of lysis buffer (174 mM ammonium chloride, 20 mM sodium bicarbonate, and 1 mM EDTA). White blood cells (WBCs) suspended in Hank's buffered salt solution containing 0.1% bovine serum albumin were loaded with 1 mM EDTA, followed by 1 unit/µL catalase (Roche Diagnostic Cooperation, Indianapolis, IN, U.S.A.) and 100 mM DHR (Molecular Probes, Eugene, OR, U.S.A.). PMA (Sigma-Aldrich, St. Louis, MO, U.S.A.) was added to the cells at a final concentration of 20 ng/mL, and the cells were incubated for 15 min. After incubation, the cells that had formed rhodamine by oxidizing DHR were counted with an EPICS® XL flow cytometer (Coulter, Miami, FL, U.S.A.).
Western blot analysis of the NADPH oxidase components
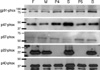
WBCs were solubilized in a lysis buffer containing 100 mM KCl, 10 mM NaCl, 10 mM HEPES, 1 mM EDTA (pH 7.4), 0.1 mM DTT, 1 mM PMSF, 10 µg/mL chymostatin, 1 µg/mL protease inhibitor cocktail (Sigma-Aldrich), 1% Triton X-100, 1% deoxycholic acid, 1% NP-40, and 0.05% SDS. The protein content of the cell lysate was resolved by 5-20% SDS-PAGE and electroblotted onto nitrocellulose membranes. Following a blocking step, the membranes were incubated for 2 hr with the following primary antibodies diluted 1:1,000: polyclonal anti-p22-phox (FL-195), anti-p47-phox (H-195), anti-p67-phox (H-300), and anti-p40-phox (D-8) (Santa Cruz Biotechnology, Santa Cruz, CA, U.S.A.); and anti-gp91-phox (Upstate, Lake Placid, NY, U.S.A.). After multiple washing steps, the membranes were incubated with HRP-conjugated anti-rabbit or anti-mouse IgG (1:10,000 dilution; Vector, Burlingame, CA, U.S.A.). Immunoblot signals were visualized with an Enhanced Chemiluminescence Detection System (Pierce Biotechnology, Rockford, IL, U.S.A.).
Isolation of total RNA and RT-RCR
Total RNA was extracted from WBCs using an RNeasy® mini kit (QIAGEN, Valencia, CA, U.S.A.), following the manufacturer's instructions. First-strand cDNA was synthesized from 2 µg of RNA with 25 ng/mL Oligo dT (Promega, Madison, WI, U.S.A.), 0.5 mM dNTP mix, 0.2 unit/µL Omniscript (QIAGEN), and 0.5 unit/µL RNase inhibitor. The reaction was incubated at 37℃ for 60 min. The synthesized cDNA was amplified using primers (Forward, 5'-ATG GGG CAG ATC GAG TGG GC-3'and Reverse, 5'-TCA CAC GAC CTC GTC GGT CA-3'; CYBA accession number, NM_000101) in a 50-µL PCR reaction containing 0.2 mM dNTP mix, 0.4 µM primer, and 1.25 units of Taq polymerase (Promega). The PCR conditions were 35 cycles of denaturation at 94℃ for 15 sec, annealing at 62℃ for 30 sec, and extension at 72℃ for 1 min. The RT-PCR products were separated in a 2% agarose gel and stained with ethidium bromide for visualization.
Isolation and amplification of genomic DNA of CYBA
Genomic DNA was extracted from peripheral blood using a Wizard® genomic DNA purification kit (Promega) according to the manufacture's protocol. The six exons of CYBA and the exon-intron junctions were amplified by PCR as previously described (13). Primers used for 5'and 3'untranslated region are as follow; 5'untranslated region: forward 5'-AAA CCA CCA AGT GCC TCG GAT G-3'and reverse 5'-TGA GCC AAT GTG GGG TTT GAG G-3', 3'untranslated region: forward 5'-CAG GCC GAC CCA GGT CCT GGC-3'and reverse 5'-CGG CCC CAG GCA GAG GCT CA-3'. PCR was performed in a 50-µL reaction mixture containing 10 pM of each primer, 0.2 mM dNTPs, 100 mM Tris-HCl, pH 8.3, 1.5 mM MgCl2, and 2.0 units of Taq polymerase (Promega). The amplification conditions were 35 cycles of 95℃ for 1 min, 59℃ for 1 min, and 72℃ for 1 min; followed by a final extension time of 5 min at 72℃. The PCR products were separated in a 1.5% agarose gel and stained with ethidium bromide for visualization.
Sequence analysis
PCR-amplified products were purified using a QIAquick PCR purification kit (QIAGEN, Hilden, Germany) and analyzed by direct sequencing in both directions using an ABI-Prism BigDye Terminator Cycle Sequencing Ready Reaction kit and an ABI Prism 3130 XL Genetic Analyzer (Applied Biosystems, Foster City, CA, U.S.A.). Primers for the sequencing reactions were the same as those used for PCR. The sequences obtained from CGD patients and healthy controls were compared with GenBank data (GenBank accession number, NM_000101) and submitted for BLAST analysis.
RESULTS
Between 1993 and 2007, 17 patients from 11 unrelated families on Jeju Island were identified as having CGD. There were 7 males and 10 females, and 5 families (45%) had multiple affected siblings. The diagnosis of CGD was made at a mean age of 2.1 yr (median, 0.5 yr; range 0.1-5.4 yr). Four patients (24%) died during the period, at a mean age of 4.7 yr (range, 0.9 to 14.3 yr). Judging from this study, the prevalence of CGD on Jeju Island is 23.1 in 1,000,000 individuals (Statistics from Jeju Special Self-Governing Province, 2007).
Among the CGD patients, 12 patients from 10 families were analyzed by DHR flow cytometric assay and Western blot analysis (Table 1). The ages of the patients at the time of genetic analysis ranged from 3.3 to 28.2 yr. With the DHR flow cytometric assay, all of the patients showed an absence of ROS production, but all other family members with normal phenotype did not show any impairment compared with healthy controls (Fig. 1). Western blot analysis demonstrated the absence of p22-phox expression in all patients, while the expression of gp91-phox, p47-phox, p67phox, and p40-phox was normal in all patients (Fig. 2). Thus, all the patients exhibited the A22° biochemical phenotype of the disease.
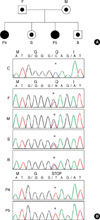
The mRNA of CYBA gene was reverse transcribed as described above. The cDNA for p22-phox in all patients and family members did not differ in size from that in healthy controls (Fig. 3). Sequence analysis of genomic DNA for CYBA gene revealed that all patients had an identical, homozygous, single-base substitution of C to T in exon 1 (c.7C>T), which was expected to result in a nonsense mutation (p.Q3X). The genomic DNA sequencing results for both parents of all the patients showed a double signal at the same position, which indicated they were heterozygous for the mutation in the CYBA gene (Fig. 4). This mutation has been previously reported at the same position in Japanese patients (14, 15).
In addition to the mutation described above, six different single nucleotide substitutions (SNP) within the CYBA gene were detected in all patients in the same manner during the course of genetic analysis (Table 2).
DISCUSSION
Jeju is a large island off southwest of Korean peninsula. The total population of Jeju Island is 563,388 (Statistics of Jeju Special Self-Governing Province, 2007). Mt. Halla rises in the center of Jeju Island, dividing it into two geographically separated regions, Jeju City and Seogwipo City. In this study, 17 patients from 11 unrelated families on Jeju Island were identified as having CGD; 14 patients were from Seogwipo City and three from Jeju City. The overall prevalence of CGD on Jeju Island is 23.1 in 1,000,000 individuals. Although we tried to identify as many patients as possible, this may be an underestimation of the actual prevalence of CGD on Jeju Island.
We hypothesized that the high prevalence of CGD on Jeju Island is associated with an identical mutation inherited from a common ancestor or proband. According to the studies carried out to date, the most common form of CGD is the X-linked form. The approximately 30-40% of CGD are inherited in an autosomal recessive traits. One of the rarest forms of CGD is caused by mutations in CYBA gene, accounting for only 5% of CGD patients (6). Up to the date, 27 mutations in CYBA gene are listed in the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/ac/all.php) and all 27 mutations reported for CYBA gene showed the allelic heterogeneity with no preponderance of common affected alleles or hot spots (16). However, all CGD patients on Jeju Island had an identical, homozygous mutation in the CYBA gene and six different SNPs within the CYBA gene in the same manner. Moreover, one of the reported Japanese patients with p22-phox-deficient CGD, sharing the same mutation with CGD patients on Jeju Island, has consanguineous parents (14). In regions with a higher incidence of consanguinity in the population, there is an increase in the prevalence of autosomal recessive genetic disorders, including CGD, as a direct result of a higher rate of inbreeding (10, 11). Altogether, these results strongly suggest that there might have been unique marriage patterns similar to endogamy or consanguineous marriage on Jeju Island.
In contrast to previous reports (13-16), a normal level of gp91-phox protein expression was detected in all CGD patients on Jeju Island. By using transgenic expression of gp91-phox and p22-phox in monkey kidney COS7 cells, which lack endogenous p22-phox and gp91-phox expression, gp91-phox was expressed on the cell surface in the absence of p22-phox expression. However, co-expression of gp91-phox and p22-phox were required to support the generation of oxygen radicals in a cell-free NADPH oxidase assay (17). This implies that the association of p22-phox with gp91-phox may be essential for regulation of electron transfer in the redox cycle, but p22-phox may not be necessary for the expression of gp91-phox protein. Further studies are required to understand the expression of gp91-phox in p22-phox-deficient CGD.
Based on different clinical parameters, X-linked CGD has a different, more severe, clinical phenotype than the autosomal recessive CGD (6). The prognosis of X-linked CGD and p22-phox-deficient CGD was not better than that of p47-phox-deficient CGD (10), and p22-phox-deficient CGD with the young age at diagnosis appears to be as severe as X-linked CGD (16). Although genetic study has not been performed, X-linked CGD had a significantly earlier age of symptom onset and age at diagnosis than autosomal recessive CGD in Korea (18). To our knowledge, there were only 2 reports about the genetic analysis of X-linked CGD in Korea (19, 20). It is hard to evaluate the clinical course of CGD patients on Jeju Island in comparison with the different genotype. However, the clinical course of CGD patients on Jeju Island seems to be considered mild in that 30% of the living patients are 20 yr of age or older.
There is an increase in the prevalence of autosomal recessive genetic disorders in regions with a higher incidence of consanguinity in the population, as a result of a higher rate of inbreeding. All the CGD patients tested on Jeju Island had an identical and homozygous mutation in the CYBA gene. This result is specific to our cohort, as the most common form of CGD in other reports is the X-linked form of CGD. Even though this study is not a nationwide survey of CGD in Korea, the finding of an identical mutation in a single region with a highly prevalent rate is quite unique. This is not consistent with the generally accepted principle of recessively inherited rare diseases. In Korea, there is a law prohibiting marriage between people with the same surname and the same family origin. However, since Jeju Island has been a geologically isolated region for a long time, there might have been unique marriage patterns similar to endogamy or consanguineous marriage. This may explain the unusual identical mutation of the CYBA gene and the high prevalence of autosomal recessive forms of CGD on Jeju Island. We are preparing to investigate the genotyping further by using microsatellite markers. This approach may help to elucidate the genetic characteristics of the population by revealing a particular familial structure on Jeju Island.
In this study, six SNPs within the CYBA gene were detected in the course of genetic analysis. Although we did not check for these SNP sites in a larger scale population, two SNPs in the CYBA gene have not yet been registered as a polymorphism in the database of SNPs (http://www.ncbi.nih.gov/SNP/): one located at nucleotide position G-36>A, which changes 13 Q codon GGG into the alternative Q codon GGA; and the other at nucleotide position 646 C>T in the 3'untranslated region of the CYBA gene.
In conclusion, we demonstrate all patients of CGD in Jeju Island have an identical, homozygous mutation in the CYBA gene.




 XML Download
XML Download