

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cystic fibrosis (CF; Mckusick 219700) is a lethal disease, which is induced by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7q31 (1). Although CF is one of the most frequent autosomal-recessive disorders in Western countries (1/2,000-1/3,000) (2), it is extremely rare in the Asians (1 in 350,000 in Japanese population) and only a few cases of CF were confirmed by mutation analysis in Korea (3-5).
The clinical features of CF are variable and depend on age at diagnosis, supportive care and treatment. The diagnosis of CF is suspected by the presence of typical clinical features such as meconium ileus at birth, recurrent or persistent pneumonia, malabsorption causing failure to thrive and growth problems, salt-wasting syndromes or obstructive azoospermia (6). The demonstration of a high sweat chloride on repeated measurements or by identifying CF mutations in two alleles could confirm the diagnosis of CF. Since CF is an extremely rare disease in Asians, sweat chloride test and neonatal screening test has not been available in Korea and there is a risk of underdiagnosis or delayed diagnosis of CF even in suspected cases.
Recently, we experienced a Korean infant with CF, who was diagnosed by genetic analysis of the CFTR gene. We present this case with a brief review of the literatures.
CASE REPORT
A 4-month of age boy was brought to emergency room at Asan Medical Center because of respiratory difficulty. In his past medical history, he was born with birth weight of 3.4 kg at the 39st week of gestation and he was the first child born to non-consanguineous parents. Delay of meconium passage and abdominal distension developed just after birth. He was diagnosed as meconium ileus and ileal resection was performed at his 5th day. Until 4-month old, he had admitted twice because of respiratory difficulty and treated as acute bronchiolitis. He was fed breast milk and gained body weight slowly up to 5.2 kg (3-10th percentile).
At 4-month of age, the respiratory symptoms such as cough, wheezing and respiratory difficulty recurred, and he was referred to Asan Medical Center. His respiratory rate was 56 breaths per minute and he exhibited 87% oxygen saturation in room air. Upon the findings of physical examination, diffuse crackles and wheezing were heard throughout entire lung field and the liver was palpated as 4 cm widths below the right subcostal margin. There was no evidence of exocrine pancreatic insufficiency and malabsorption yet. Hypoalbuminemia (2.5 g/dL), elevated liver enzyme (AST/ALT: 59/42 IU/L) and mild jaundice (total bilirubin/direct bilirubin: 4.3/2.5 mg/dL) were detected.
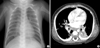
Chest radiographs, computed tomography scan of the chest revealed multifocal atelectases and peribronchial infiltration (Fig. 1). No respiratory tract anomaly was found by bronchoscopic examination. Moderate fatty liver and mild hepatomegaly was detected by abdominal ultrasonography. Oxygen supplementation via nasal prong (2-5 L/min) and supportive care including bronchodilator therapy were started. But his breathing became more difficult at 8th hospital day, and he was placed on artificial ventilation therapy for 28 days. Staphylococcus aureus and Stenotrophomonas maltophilia were cultured from his bronchial secretion. Tests for TORCH infection, causing neonatal cholestasis and lung infiltration, were all negative.
From his past medical history and clinical course, CF was suspected. The sweat chloride test could not be performed due to his poor medical condition. To confirm the diagnosis of cystic fibrosis, direct sequencing analysis of all coding exons and the exon-intron boundaries of CFTR gene was performed. Used primer sequences are listed in Table 1. He was a compound heterozygote of two mutations: c.263T>G (p.Leu88X) in exon 3 from his father and c.2089_2090insA (p.Arg697-LysfsX33) in exon 13 from his mother (Fig. 2). Both of them belong to the mutation class I, which is most severe group of CFTR mutations (7). The c.263T>G (p.Leu88X) has been reported in a Korean CF patient already (8), and the c.2089_2090insA (p.Arg697LysfsX33) was a novel frameshift mutation.
He recovered by artificial ventilation, mucolytics, antibiotics and active physiotherapy. Hypoalbuminemia, cholestasis and hepatomegaly were improved slowly with sufficient feeding, supplementation of pancreatic exocrine enzymes, multi-vitamins and essential fatty acid. He is 19-month old now and has admitted to the hospital at the age of 12 months because of hyponatremic dehydration with metabolic alkalosis following to acute gastroenteritis. However, he shows catch-up growth and his height and weight is with in 10-25th percentiles with supplementation of pancreatic exocrine enzymes and multi-vitamins (Fig. 3).
DISCUSSION
The causing gene of CF is the CFTR gene, which is located on chromosome 7q31. The gene spans 250 kilobases, encoding 1,480 amino acids in the mature protein. CFTR protein has been shown to function as a regulated chloride channel, which in turn may regulate the activity of other chloride and sodium channels at the cell surface (1, 9). More than 1,200 disease-associated mutations have been described already, and the most common mutation in Western population is ΔF508, which is accounting for approximately 70% of defective CFTR alleles (9). Over 90% of CFTR gene mutations could be identified by using a panel of 70 mutations in general population of the United States (10). In Korean population, However, only 13 mutations have been reported by now (3, 5, 8, 11), and these are not frequently found mutations in Caucasians. Our patient is a compound heterozygote with a known mutation of Leu88X and a novel mutation of Arg697LysfsX33. This novel mutation is located in exon 13, which belongs to highly charged regulatory domain. Some mutations such as Glu692X and Glu695GlyfsX35 were already reported, and these are located near Arg697LysfsX33 mutation. The all patients who carried these mutations showed severe clinical phenotypes (12, 13). Both two mutations of our patient are classified as mutation Class I, which is most severe group of CFTR mutations leading to premature termination of mRNA and virtually absence of functional CFTR protein (14). Clinical consequences of the CFTR defect for various organs affected in CF patients are site-specific and range from severe to mild. Although the correlation between genotype and phenotype is not fully understood in CF, genotype is thought to be a good predictor of exocrine pancreatic function (pancreatic insufficiency vs. pancreatic sufficiency) (15). However, respiratory symptoms are poorly correlated to CFTR genotype with few exceptions (16). Meconium ileus is not genotype-dependent in the fashion characteristic for pancreatic function, but this symptom is almost exclusively observed in patients with pancreatic insufficiency and severe CFTR genotypes (17). Early and severe presentation of our patient might be caused by severe mutations and the absence of CFTR function.
In conclusion, we experienced one case of CF with a novel mutation of CFTR gene. Although CF is a rare disease in Korea, it should be considered in the differential diagnosis of any infant or child presenting with early onset recurrent bronchiolitis, especially in patients with past history of meconium ileus.



 XML Download
XML Download