

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The p63 gene is a member of the p53 gene family and it shares a high degree of homology and remarkable structural similarity with p53 gene (1, 2). Therefore, the p63 protein was originally thought to be another tumor suppressor functioning in a similar capacity to p53. However, p63 has been demonstrated to have divergent roles. It is a key transcriptional regulator of the proliferation and differentiation cascade in stratified epithelia (3), it exhibits tissue-specific roles in normal development and a complex contribution to tumorigenesis which is due to its expression as multiple protein isoforms (4). According to different amino terminals, p63 has two major isoforms: TAp63 and ΔNp63. TAp63 has a transactivating amino terminal while ΔNp63 has a dominant negative activating amino terminal (5-8). In general, the TAp63 isoform behaves like p53 because they transactivate various p53 downstream targets and induce apoptosis, mediate cell cycle control. In contrast, the ΔNp63 isoform has been shown to display functions opposite to that of the TAp63 isoform, including acting as oncoproteins (9-12). ΔNp63 overexpression is often observed in and enhances oncogenic growth of squamous cell carcinomas (11, 13). ΔNp63 could function as dominant-negative molecule against the p53 tumor suppressor activities (5). ΔNp63 overexpression induces nuclear accumulation of β-catenin and activates β-catenin signaling that promotes cell proliferation (12). Nowadays, many researchers tend to agree with the concept that TAp63 plays a more p53-like role, whereas ΔNp63 has an antagonistic or even oncogenic role in cancer progression (11, 14-17).
Our previous study demonstrated that ΔNp63 was overexpressed in human transitional cell carcinoma of bladder (TCCB) tissues at both mRNA and protein levels, so we hypothesize that ΔNp63 promotes the survival and proliferation of both epithelial stem cells and TCCB cells. However, the specific role of ΔNp63 in TCCB development remains largely unclear and the mechanisms by which ΔNp63 promotes cell survival remain to be elucidated. Understanding the regulation and mechanistic contributions of ΔNp63 in TCCB may ultimately provide new therapeutic opportunities for this disease. Thus, the objective of this study was to better understand the functions of ΔNp63 in TCCB with a human TCCB cell line 5637 in vitro and in a nude mouse xenograft model where the expression of ΔNp63 was silenced by RNA interference (RNAi) technology.
MATERIALS AND METHODS
Plasmid construction
A pair of effective short hairpin RNA (shRNA) against ΔNp63 mRNA was selected from a preliminary experiment. The pair of shRNA forms a structure consisting of two 19-bp stem targeting ΔNp63 mRNA, a 9-bp loop and a short poly(A)6 sequence. Two oligonucleotides, forward, 5'-GATCCGTGCCCAGACTCAATTTAGTTTCAAGACGACTAAATTGAGTCTGGGCATTTTTTGTCGACA-3', reverse, 5'-AGCTTGTCGACAAAAAATGCCCAGACTCAATTTAGTCGTCTTGAAACTAAATTGAGTCTGGGCACG-3', were synthesized and ligated directly into BamHI and HindIII linearized genesil-1 plasmid (Jingsai Inc, Wuhan, China). The recombinant ΔNp63-shRNA expression construct, pΔNp63-shRNA, was confirmed by using PstI+SalI double digestion and by gene sequencing. The negative control plasmid, termed pΔNp63-cRNA has an sequence insert at the same place from the following two oligonucleotides: forward, 5'-GATCCGACTTCATAAGGCGCATGCTTCAAGACGGCATGCGCCTTATGAAGTCTTTTTTGTCGACA-3'; reverse, 5'-AGCTTGTCGACAAAAAAGACTTCATAAGGCGCATGCCGTCTTGAAGCATGCGCCTTATAAGTCG-3'.
Cell culture and transfection
Human TCCB cell line 5637 was purchased from the Institute of Cell Research, Shanghai, Chinese Academy of Sciences. The cells were cultured in RPMI 1640 medium (Gibico, Shanghai, China) supplemented with 10% fetal bovine serum (FBS, Sijixin Inc., Beijing, China) and 1% penicillin-streptomycin (Invitrogen, Shanghai, China). All cells were cultured at 37℃ with 5% CO2. For transfection, cells were seeded in 6-well plates at 1 × 106 cells per well and allowed to grow overnight to approximately 80% confluence. For each well, cells were transfected with the mixture of 0.4 µg pΔNp63-shRNA or pΔNp63-cRNA or 0.4 µL phosphate-buffered solution (PBS) and 10 µL Effectene transfection reagent (Qiagen, Shanghai, China) in 600 µL fresh 1640 culture medium. Forty-eight hours after transfection, cells were harvested for immunocytochemistry, reverse transcription polymerase chain reaction (RT-PCR) or flow cytometry analysis as described below.
Immunocytochemistry
Cells were fixed with pre-chilled acetone at 4℃ for 30 min, then incubated for 10 min with 3% hydrogen peroxide in methanol. Goat non-immune serum was used to block the non-specific binding. Cells were incubated overnight at 4℃ with a 1:200 dilution of mouse monoclonal anti-ΔNp63 antibody (Santa Cruz, Shanghai, China) or with a 1:100 dilution of rabbit anti-cyclin D1 polyclonal antibody (Santa Cruz). Then cells were washed three times and an appropriate biotinylated-conjugated secondary antibody (Santa Cruz) with a 1:150 dilution was applied for 15 min at 37℃. Cells were washed three times and then incubated with streptavidin peroxidase (SP, Zhongshan Golden Bridge Inc., Beijing, China) for 15 min at 37℃. Then horseradish peroxidase substrate diaminobenzidine solution was then added for 15 sec and the staining was stopped by washing the coverslip with water repeatedly. Finally, cells were counterstained with hematoxylin.
Semiquantitative multiplex RT-PCR
Total RNA was extracted from tissue homogenates or cell lysates with TRIzol reagent (Invitrogen) and RT-PCR was carried out with a RNA PCR Kit Ver.3.0 (TaKaRa, Dalian, China) according to the kit's instructions. The primers used for ΔNp63 were: forward, 5'-TGCCCAGACTCAATTTAGTGAG-3'; reverse, 5'-TCTGGATGGGGCATGTCTTTGC-3', which yields a product of 335 bp. The primers used for β-actin were: forward, 5'-GTGGACATCCGCAAAGAC-3'; reverse, 5'-AAAGGGTGTAACGCAATCAA-3', which yields a product of 302 bp. The PCR condition was: 94℃ for 2 min, then 35 cycles at 95℃ for 1 min, 53℃ for 0.5 min, and 72℃ for 1 min in 1.5 mM MgCl2-containing reaction buffer. Five microliters of RT-PCR products were resolved on 1.5% agarose gels. The gels were stained with ethidium bromide (EB) and were scanned for densitometric estimation of the ΔNp63 products with β-actin products serving as the internal control.
Cell cycle assay with flow cytometry
A total of 1 × 106 cells treated with pΔNp63-shRNA vector or pΔNp63-cRNA vector were trypsinized and washed with PBS twice. Then cells were transferred and fixed in prechilled 75% ethanol at 4℃ overnight. After three times of wash, cells were digested with 1% RNase at 37℃ for 30 min and stained with 50 µg/mL propidium iodide (PI, Sigma, Shanghai, China) for 1 hr at 4℃. Samples were assayed by a Coulter Epics XL flow cytometer (Beckman-Coulter Inc., NewYork, NY, U.S.A.) and data were analyzed with Multicycle DNA content and cell analysis software.
3-(4, 5-Dimethylthiazol-2-yl)-2, 5-dimethyl tetrazolium bromide (MTT) assay for cell proliferation
Cells in logarithmic growth phase were seeded in 96-well plates at 2 × 104 cells per well. Then cells were transfected with pΔNp63-shRNA or pΔNp63-cRNA or PBS and continued to culture for 24, 48, 72, 96, and 120 hr respectively. Four hours before stop culturing, 20 µL of 5 mg/mL MTT (Sigma) was added to the culture medium. After incubation, the culture medium was removed and 200 µL of dimethylsulphoxide (DMSO) was added to resolve the crystal. Absorbance was measured at 490 nm. Each sample was assayed for four times.
Nude mouse xenograft model
Female BALB/c nude mice, 4 weeks of age, weighting 13±0.5 g, were purchased from Experimental Animal Center of Chinese Academy of Sciences, Shanghai, China. Mice were housed in microisolator cages in a specific pathogen-free (SPF) condition with 12-hr light-dark cycles. Mice were subcutaneously implanted with 2 × 106 5637 cells. Once tumors reached approximately 60 µL in volume, the mice were allocated to receive either pΔNp63-shRNA or pΔNp63-cRNA vector or PBS. Complexes of 20 µg vector (or PBS)+4 µL in vivo-jetPEI™ (polyplus-transfection Inc., Shanghai, China)+160 µL 5% glucose were directly injected into the tumor once a week for five weeks. Tumor dimensions were measured weekly and the tumor volumes calculated using the formula: [1/2] × a × b2, where a and b, respectively, represented the larger and smaller tumor diameter. At the end of the five week treatment, mice were killed by overdose of ketamine (400 mg/kg) and xylazine (50 mg/kg) and necropsy was performed. Tumor tissue samples were prepared for immunohistochemistry or RT-PCR or transmission electron microscopy. Tumor growth inhibition (TGI) was calculated using the formula TGI (%)=(1-MT/MC) × 100, where MT and MC are the mean tumor masses in the treatment group, and control group, respectively.
Morphologic and immunohistochemical assessment of apoptosis in nude mouse xenograft model
For histological examination, tumor tissue samples were fixed with 4% paraformaldehyde for 72 hr, dehydrated in graded ethanol, and embedded in paraffin. Samples were cut into 5-µm-thick sections and stained by hematoxylin-eosin (HE).
For transmission electron microscopy, tumor tissue samples were cut into sections approximately 1 × 1 mm, fixed with 4% paraformaldehyde for 2 hr, and then transferred into pre-chilled 1% glutaraldehyde. The samples were dehydrated in graded ethanol, embedded in epon 812, and then cut into ultrathin or semithin sections. The sections were stained and examined under a Hitachi-800 transmission electron microscope.
For tumor tissue immunohistochemistry, sections of 5-µm-thick paraffin-embedded tissue microarrays were deparaffinized and rehydrated with xylene and ethanol. The immunostaining procedures were the same with the immunocytochemistry described above.
RESULTS
ΔNp63 mRNA and protein expression were inhibited by ΔNp63 shRNA in vitro and in vivo
As previously reported (18), TCCB cell line 5637 inherently expresses high levels of ΔNp63. Following transfection of pΔNp63-shRNA, at 48 hr later, the relative ΔNp63 mRNA expression levels (ΔNp63/β-actin) were 0.156±0.07 for cells transfected with pΔNp63-shRNA, 0.749±0.02 for cells transfected with the control vector, and 0.786±0.04 for cells transfected with PBS. Compared with the control RNA vector, the expression levels of ΔNp63 mRNA were significantly inhibited by ΔNp63 shRNA (p<0.05) (Fig. 1).
The expression levels of ΔNp63 protein were detected at 48 hr post-transfection by immunocytochemistry. ΔNp63 proteins positive cells should be stained brown in the nuclei. Cells transfected with PBS or the control vector were strongly stained in the nuclei (Fig. 2A, B), while cells transfected with pΔNp63-shRNA were only weakly stained in the nuclei (Fig. 2C). These demonstrated that ΔNp63 shRNA inhibited ΔNp63 protein expression significantly.
Similar suppressions of ΔNp63 mRNA and protein expression by ΔNp63 shRNA were found in the nude mouse xenograft model by semiquantitative RT-PCR and immunohistochemistry.
ΔNp63 shRNA caused cell cycle arrest of 5637 cells in vitro
Cell cycling of 5637 cells was assessed by flow cytometry. The percentage of cells in G0/G1 phase for cells transfected with pΔNp63-shRNA, pΔNp63-cRNA or PBS was respectively 66.38±3.08%, 52.58±2.03% and 48.27±0.84%. The percentage of cells in S phase for the three groups was 33.68±2.06%, 39.82±1.89% and 40.50±0.58%, respectively (Fig. 3). These results indicated that a significant percentage of 5637 cells were arrested in G0/G1 phase after treatment of ΔNp63 shRNA.
ΔNp63 shRNA inhibited 5637 cell proliferation in vitro
Proliferation of 5637 cells was evaluated with the MTT assay (Fig. 4). The absorbance-time curves showed that the absorbance of cells transfected with pΔNp63-shRNA was significantly lower than that of cells treated with the control vector or PBS. The difference is especially significant at 120 hr post-transfection. The result indicated that the proliferation of TCCB cell line 5637 was significantly inhibited by ΔNp63 gene silencing.
ΔNp63 expression suppression led to down-regulation of cyclin D1 in vitro and in vivo
The effects of ΔNp63 gene silencing on the expression of cyclin D1 was evaluated by immunochemistry staining which was performed both in 5637 cell line in vitro and in nude mouse xenograft tumor. Cells with positive cyclin D1 protein expression should be stained brown in the nuclei. Both in 5637 cells in culture (Fig. 5) and in its nude mouse xenograft tumor (Fig. 6), cells transfected with pΔNp63-shRNA were stained weakly positive in the nuclei, while cells treated with PBS or the control vector were stained strongly positive in the nuclei. These results suggested that silencing of ΔNp63 expression is associated with downregulation of cyclin D1 expression in 5637 cells.
ΔNp63 shRNA transfection inhibited 5637 tumor growth in vivo
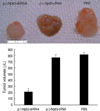
Tumor volumes were determined weekly. At the end of the experiment, mice receiving pΔNp63-shRNA exhibited a 74.44% reduction in final tumor size relative to mice treated with pΔNp63-cRNA and tumor-bearing mice treated with PBS had a similar tumor size as the control shRNA group (Fig. 7). Neither group exhibited signs of systemic toxicity or metastasis.
Morphologic changes in tumor
Histological assessment revealed that, compared to control shRNA and PBS treated mice, more inflammatory cells and sometimes zones of necrosis were found in tumor tissues of mice treated with pΔNp63-shRNA (data not shown). Transmission electron microscopy indicated that disintegration of nucleolus, vacuoles in cytoplasm, inflammatory cell infiltration and apoptotic body formation could be observed in tumor tissues treated with PΔNp63-shRNA (Fig. 8A, B) which were rarely seen in tumor tissues treated with the PΔNp63-cRNA or PBS (Fig. 8C, D)
DISCUSSION
The ability of RNAi mediated by shRNA to silence individual gene expression with a high degree of specificity presents a unique opportunity to study gene functions (19, 20). Plasmid and viral vectors producing shRNA using the polymerase III promoter offer efficient and stable gene silencing (21, 22). In this study, we constructed a ΔNp63 shRNA expression plasmid, which achieved efficient and specific ΔNp63 gene silencing in human TCCB cell line 5637 following transfection both in vitro and in a nude mouse xenograft model. With ΔNp63 gene silencing, the cell cycle of 5637 cells was arrested in G0/G1 phase and cellular proliferation was significantly suppressed. ΔNp63 gene silencing by specific shRNA in the nude mouse xenograft tumor resulted in marked retardation of tumor growth and increase in tumor cell apoptosis. Our data suggested that ΔNp63 might play an oncogenic role in TCCB progression through promoting cell survival and proliferation. Therefore, ΔNp63 might be a new target gene for TCCB gene therapy, and ΔNp63 shRNA might be a novel and promising approach for silencing ΔNp63 expression.
One interesting new finding in our study is that cyclin D1 expression was downregulated after silencing of ΔNp63 expression in 5637 cells both in vitro and in vivo, indicating cyclin D1 might be a target gene of ΔNp63. ΔNp63 shares homology with the DNA-binding domain of p53, and numerous studies have reported that ΔNp63 proteins could bind to p53 consensus DNA sequences (23, 24). Cyclin D1 had previously been identified to have a p53 binding site. Cyclin D1 is a key regulator of cell cycle progression and a proven oncogene in several cancers, including urinary bladder carcinoma (25). It regulates cell cycle progression by activating cyclin-dependent kinases 4 and 6 (CDK4 and CDK6), which in turn phosphorylates the retinoblastoma protein (Rb), leading to the inactivated pRB, releasing of transcription factor E2F and cell progression through G1/S checkpoint (26-30). The strong positive correlation between ΔNp63 and cyclin D1 expression found in our study suggests that ΔNp63 might regulate TCCB cell cycle through regulating the expression of cyclin D1 which leads to less efficient phosphorylation of Rb which is a key condition for cells to pass G1/S regulation point (31, 32). Therefore, cyclin D1 downregulation could lead to cell cycle arrest in G0/G1 phase, inhibition of cellular proliferation and promotion of apoptosis.
In conclusion, this study demonstrated that ΔNp63 plays an oncogenic role in TCCB cells. Targeting this oncoprotein using a shRNA expressing vector induced specific silencing of the gene. Suppression of ΔNp63 expression impaired TCCB tumor growth in vivo and improved the survival of tumor-bearing nude mice. These effects are associated with impaired cellular proliferation and increased apoptosis. At the molecular level, our study suggests that cyclin D1 might be one of the target genes of ΔNp63.







 XML Download
XML Download