

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Klebsiella pneumoniae is a relevant opportunistic pathogen that accounts for up to 10% of all nosocomial infections (1, 2). It is a frequent causal agent of urinary tract infections, septicemia, and pneumonia in immunocompromised patients, and it is also an important pathogen for community-acquired infections. Its importance in clinical settings is increasing due to its ability to produce extended-spectrum β-lactamase (ESBL) (3). It has been reported that ESBL-producing organisms tend to be multidrug-resistant and they have an increased risk of treatment failure (3, 4). Thus, it is indispensable to detect these ESBL-producing isolates and to control their emergence and spread.
Several molecular methods have been used for characterizing K. pneumoniae isolates such as randomly amplified polymorphic DNA analysis, pulsed-field gel electrophoresis, and amplified fragment length polymorphism (5-7). However, these methods are band-based and so the results may be ambiguous, but generally these methods are well suited for local epidemiologic investigation. Yet, it is very difficult to compare the results generated at different laboratories for conducting a global epidemiologic analysis. To overcome the limitations of the band-based typing methods, multilocus sequence typing (MLST) has been developed, which is a nucleotide sequence-based method for bacterial or fungal typing (8-10). It is highly discriminative and it is easy to standardize, store, and exchange information electronically. It has been applied successfully for the epidemiologic characterization of a variety of clinically important bacterial pathogens, such as Streptococcus pneumoniae, Staphylococcus aureus, Enterococcus faecium, Neisseria meningitidis, and Campylobacter jejuni (9-13). Very recently, a MLST method for K. pneumoniae by using seven housekeeping genes, such as rpoB, gapA, mdh, pgi, phoE, infB, and tonB, has also been developed (14).
We developed another MLST scheme for K. pneumoniae isolates based on five housekeeping genes, and we characterized the ESBL-producing K. pneumoniae isolates from an outbreak in a Korean hospital.
MATERIALS AND METHODS
Bacterial isolates
From August 2004 to May 2005, a total of 66 ESBL-producing K. pneumoniae were isolated from patients in an intensive care unit (ICU) of the Kangbuk Samsung Hospital (Seoul, Korea), which is a tertiary care hospital with 645 beds. Of these, 43 isolates were preserved and included in this study (Table 1). Twenty-nine isolates were from sputum, and 11 isolates were from urine; the others were isolated from blood or bile. Thirty-eight K. pneumoniae isolates, which were isolated at another three Korean tertiary hospitals as a part of a surveillance study that was conducted in 1999 and 2004, were also investigated in this study for comparisons (Table 2). Thirty-one isolates were collected from the Samsung Medical Center (Seoul, Korea), and others were from the Dong-A University Hospital (Busan, Korea) and the Chungbuk National University Hospital (Cheongju, Korea). Of these 38 surveillance isolates, 15 produced ESBL and the others did not. For screening for ESBL, the GNS-650 card (Vitek system, bio-Merieux, Hazelwood, MO, U.S.A.), which is based on the use of cefotaxime and ceftazidime with or without clavulanic acid, was used. For a reference, K. pneumoniae ATCC 13883 was also analyzed. Thus, a total of 82 K. pneumoniae isolates were analyzed.
Multilocus sequence typing
We selected five housekeeping genes to establish the MLST scheme; rpoB (RNA polymerase β-subunit), gyrA (DNA gyrase subunit A), gapA (glyceraldehydes 3-phosphate dehydrogenase A), groEL (GroEL protein), and gyrB (DNA gyrase subunit B). The primers we used for amplification of the gene fragments are shown in Table 3. PCR amplifications were carried out under the following conditions: 35 cycles of denaturation at 94℃ for 30 sec, annealing at 50-55℃ for 30 sec, and extension at 72℃ for 1 min; this was all preceded by a 5 min denaturation at 94℃ and then it was all followed by a 5 min final extension at 72℃. The PCR products were purified using a PCR purification kit (Bioneer, Daejeon, Korea), according to the manufacturer's recommendations, and then the products were sequenced on an ABI3700 DNA sequencer (Applied Biosystems, Foster City, CA, U.S.A.).
The raw sequences were concatenated and edited by using the EditSeq and MegAlign programs (DNASTAR, Madison, WI, U.S.A.). For each locus, distinct allele sequences were assigned as an arbitrary allele number. Each isolate was characterized by its allelic profile, which was represented as a series of 5 integers corresponding to the alleles at each of the loci, in the order of rpoB, gyrA, gapA, groEL, and gyrB. The sequence type (ST) was designated for each unique allelic profile. Clusters of related STs sharing three or more identical alleles out of the five were defined as clonal complexes (CCs), which were determined by the eBURST program (http://eburst.mlst.net/loci.asp) (15). The dN/dS ratios, with dN being the number of nonsynonymous substitutions per nonsynonymous site and dS being the number of synonymous substitutions per synonymous site (16), were determined by using the START program (http://outbreak.ceid.ox.ac.uk/software.htm). The index of association (IA) was also calculated with the START program to test to what extent the K. pneumoniae population was clonal (17). IA is a measure of the variance in the number of allelic mismatches relative to that expected under the hypothesis of panmixia (random association), where the mean number of pairwise allelic mismatches is a measure of genetic distance. When the IA value does not deviate significantly from zero, the alleles are in linkage disequilibrium, i.e., there is frequent recombination or non-clonal population structure.
Pulsed-field gel electrophoresis (PFGE)
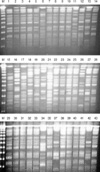
The 43 ESBL-producing K. pneumoniae isolates recovered from an outbreak at the Kangbuk Samsung Hospital were characterized by PFGE as described elsewhere (18, 19). Agarose plugs containing genomic DNA were digested with SmaI (Gibco, BRL, Gaithersburg, MD, U.S.A.), according to the manufacturer's recommendations. The DNA restriction fragments were separated with using a CHEF-Mapper apparatus (Bio-Rad Lab.), at 6 V/cm for 22 hr. The PFGE patterns were interpreted with using the published criteria (20). A divergence in more than three bands was interpreted as indicative of different PFGE types.
Detection of bla genes
The bla genes related to ESBL enzymes were assayed by PCR with the corresponding primers for the TEM, SHV, CTX-M, and OXA ESBL types, as described previously (21). To specify the subtype of the bla genes, the amplified PCR products were sequenced on both strands as was done in the MLST analysis. The amino acid sequences were deduced from the nucleotide sequences using the MegAlign program (DNASTAR), and they were compared with the database of the website (http://www.lahey.org/Studies/).
RESULTS
MLST scheme for K. pneumoniae isolates
Sequence data were obtained for the five selected loci from all 82 K. pneumoniae isolates included in this study. The nucleotide sequences at each locus ranged from 271 bp (gapA) to 752 bp (gyrA). The number of different alleles ranged from 9 (gapA) to 20 (gyrA). The proportion of sites at which nucleotide variation was observed within each locus that was examined ranged from 3.93% (rpoB) to 8.49% (groEL) (Table 4). By calculating the dN/dS ratio, the degree of selection operating on each locus was evaluated. No nonsynonymous substitutions were found in the gapA and gyrB loci, so the dN/dS ratios could not obtained for them. The dN/dS ratios for the other loci were significantly less than 1 (Table 4), indicating there was no strong positive selective pressure on the selected genes. Thus, these five loci could be appropriate for the K. pneumoniae MLST scheme. The index of association (IA) was used to test for linkage disequilibrium between the alleles at the five loci. The IA value was 1.951 for all the K. pneumoniae isolates. This value was reduced to 0.524 when including only one representative isolate for each sequence type (STs), and this reflects the moderate linkage equilibrium or clonal population structure of the K. pneumoniae isolates included in this study.
Based on the nucleotide variations of the five genetic loci, a total of 37 different STs could be identified among 82 K. pneumoniae isolates (Table 1, 2). The majority of these (29 out 37 STs, 78.4%) were represented by single isolate. Among the STs shared by multiple isolates, the most frequently encountered were ST2 (25 isolates, 30.5%), ST20 (8 isolates, 9.8%), and ST15 (5 isolates, 6.1%). Other STs (ST9, -11, -13, -14, and -21) represented two to four isolates. These STs were grouped, using the eBURST program, into CCs that consisted of all the isolates of the STs that shared three or more identical alleles (Fig. 1). As a result, six CCs were identified among the 82 K. pneumoniae isolates. CC20 included six STs (ST17, -18, -20, -21, -26, and -32), of which ST20 was assumed as a founder. Fourteen isolates (17.1%) were included in the CC20. CC2 included four STs (ST2, -13, -15, and -37), of which ST2 was assumed as a founder. Thirty-four isolates (41.5%) belonged to the CC2, but most of them were isolated at the Kangbuk Samsung Hospital during an outbreak. CC36 also included 7 isolates (8.5%) of four STs (ST4, -9, -31, and -36), of which ST36 was assumed as a founder. The other three CCs each consisted of two STs (Table 1, 2, Fig. 1).
PFGE and MLST
PFGE was performed for 43 outbreak isolates from Kangbuk Samung Hospital. Among the 43 outbreak isolates, seven PFGE types (A to G) were identified (Fig. 2 and Table 1). The PFGE patterns were well concordant with the STs in the MLST. Two subtypes, AA and DD, could be differentiated from A and D, respectively. All 31 isolates of the PFGE type A belonged to CC2, which were differentiated into ST2 or ST15. ST2 and ST15 were single-locus variants of each other, differing at rpoB. One isolate (KS412-6) of PFGE type AA belonged to ST13, a single-locus variant of ST2 with a different allele in gyrA. PFGE type B was correlated with ST36 of CC36. Isolates of the PFGE type D and DD showed ST11 and ST14, respectively, both of which belonged to CC11-14. Both isolates of the PFGE type G belonged to CC20, but they could be differentiated into ST17 and ST18, which are different at gyrA. The other three PFGE types, C, E, and F, represented single isolates.
ESBL-producing K. pneumoniae isolates with outbreak
Of the 43 ESBL-producing K. pneumoniae isolates recovered from the Kangbuk Samsung Hospital, all but one produced SHV enzymes (29 isolates of SHV-1, 5 of SHV-5, 8 of SHV-11, and one each of SHV-12 and SHV-14). All but four isolates produced CTX-M-14 and 10 isolates were positive to blaTEM-1 (Table 1). One isolate (KS412-4) was negative on PCR assays for the four bla genes tested in this study.
All 33 outbreak isolates of CC2 and PFGE type A produced SHV and CTX-M enzymes. All the isolates produced CTXM-14, and all but three isolates produced SHV-1. The other three isolates produced SHV-5. Nine K. pneumoniae isolates produced TEM-1. Except for one isolate (KS502-5) of CC3-16 and PFGE type F, all the TEM-1-producing isolates belonged to CC2 and they showed PFGE type A. Four isolates of CC36 and PFGE type B produced SHV-11 and CTX-M-14. Of the three isolates of CC11-14, two of ST11 and the PFGE type D produced SHV-11 and CTX-M-14, while one of ST13 and PFGE type DD produced SHV-1 and CTX-M-14. Two isolates of CC20 and PFGE type G produced both SHV-11 and CTX-M-14, or only SHV-11.
K. pneumoniae isolates from other hospitals
Of the 38 K. pneumoniae isolates collected from three Korean hospitals as a part of a surveillance program, 15 isolates produced ESBL. All 15 ESBL-producing isolates produced SHV enzymes. Among these surveillance isolates, SHV-12 was the most frequent (10 isolates), and this was in contrast to the outbreak isolates, of which SHV-1 was the most common. Five isolates produced CTX-M-14, and 8 isolates were positive to blaTEM-1 (Table 2). Of the 15 ESBL-producing isolates, nine belonged to CC20, six isolates belonged to ST20, two belonged to ST21, and one belonged to ST26. There might be no obvious correlation between CC and the bla genes. Twenty-three K. pneumoniae isolates did not produce ESBL. These ESBL-negative isolates were more diverse than were the ESBL-producers. Only three isolates belonged to CC20 (ST20 and ST32), and three isolates belonged to CC2 (ST13 and ST37). Most of the isolates formed singletons in MLST analysis.
DISCUSSION
The objectives of this study were to demonstrate development of a typing method for K. pneumoniae and to investigate the characteristics of the ESBL-producing isolates recovered during an outbreak within a Korean hospital. Molecular typing is prerequisite for elucidating the epidemiology and population structure of bacterial pathogens. Out of several molecular typing methods, MLST is becoming popular owing to its several advantages over the other typing methods, which have been discussed repeatedly elsewhere; a high level of discrimination, it is unambiguous and reproducible, and its scalable nature resulting from use of nucleotide sequences, it has electronic portability via the Internet, it can easily analyze the generated data, and it has a wide applicability (9, 10, 22, 23). Very recently, the MLST scheme for K. pneumoniae was reported on and this was based on seven housekeeping genes (14). Diancourt et al. (14) characterized 67 K. pneumoniae isolates that were collected from several European countries with using their MLST scheme and they compared the results with those from ribotyping.
In this study, we presented another MLST scheme for K. pneumoniae based on five housekeeping genes. The appropriate nucleotide variations of each gene fragment (3.93% to 8.49%) indicated that these genes were applicable for the MLST scheme. The low of dN/dS ratio (0 to 0.1068), which indicated the neutral feature of the nucleotide substitution, also suggested that the selected housekeeping genes are probably suitable for population genetic studies of K. pneumoniae. Although our MLST scheme could not be compared with that of Diancourt et al. (14) because of the different loci and isolates, it showed results concordant with the PFGE. That is, the isolates with a distinct PFGE pattern belonged to a different ST. Moreover, some PFGE types could be differentiated into two STs, for example, the PFGE type A was differentiated into ST2 and ST15. Thus, our MLST procedure presented here is probably useful for a short-term epidemiologic study as well as for a global epidemiologic study, although only five loci were included in the scheme.
Using the MLST scheme, we investigated the molecular characteristics of ESBL-producing K. pneumoniae isolates from an outbreak within a Korean hospital. At the Kangbuk Samsung Hospital, at most three or less ESBL-producing K. pneumoniae had been isolated per month by July 2004. From August 2004 to May 2005, we observed an outbreak of ESBL-producing K. pneumoniae isolates primarily at the ICU. During this period, a total of 66 ESBL-producing K. pneumoniae were isolated at up to 14 isolates a month. Although we did not preserve the isolates found in August 2004, we could include the 43 ESBL-producing K. pneumoniae isolates from September 2004 to May 2005 in this study.
The results of MLST indicated that most of the isolates collected during the outbreak period belonged to the same clone, ST2 (allelic profile, 2-9-2-12-3). Particularly, all the isolates except two belonged to ST2 early during the outbreak period (September and November, 2004). After November 2004, ST2 was still the most prevalent, but its proportion decreased. In addition, five isolates emerged with its single-locus variant, ST15. ST15 (7-9-2-12-3) differed from ST2 by a single nucleotide of rpoB, which was probably due to a point mutation, and so it must have diverged from ST2 (24, 25).
Most of these outbreak isolates used in this study produced SHV and CTX-M enzymes as the ESBL enzymes (Table 1). A previous study performed in other Korean hospitals also reported that SHV and CTX-M were the most common ESBL enzymes (21). SHV-12 was the most frequently encountered SHV enzyme in the previous studies (21, 24-26), which was also confirmed in this study by the investigation of ESBL-producing K. pneumoniae isolates from Korean hospitals other than the Kangbuk Samsung Hospital (Tabel 2). However, SHV-11 was the most common among the outbreak K. pneumoniae isolates. This indicates that the outbreak K. pneumoniae isolates that produce ESBL are different from the prevailing isolates among the other Korean hospitals. However, we could not conclude whether this clone had previously existed in the Kangbuk Samsung Hospital and had spread during the outbreak period or whether it was transferred from another hospital or the community because we did not investigate the characteristics of the ESBL-producing K. pneumoniae isolates prior to the outbreak.
One of the interesting findings in this study was probably the different distribution of CCs between the ESBL-producing isolates and the non-producing isolates. Of the 12 ESBLproducing K. pneumoniae isolates from the Samsung Medical Center in 2004, eight isolates (66.7%) belonged to CC20. However, only three out of 23 ESBL-nonproducing isolates (13.0%) that were collected from the same hospital during the same period belonged to CC20 (Table 2). Although this result was based on the small number of isolates and thus it might be premature to draw a conclusion, it may suggest that the ESBL genes prefer to incorporate into a particular clone (s) as in methicillin-resistant S. aureus and vancomycinresistant E. faecium (12, 27, 28). Further investigation by including more isolates from diverse localities will be required to prove such a hypothesis.
In summary, we developed a new MLST scheme for K. pneumoniae based on five housekeeping genes. We investigated the molecular characteristics of ESBL-producing K. pneumoniae isolates from an outbreak within a Korean hospital with using the developed MLST method. Most of the outbreak isolates belonged to ST2 and they produced SHV-1 and CTXM-14, which was different from the features of the K. pneumoniae isolates from other Korean hospitals.




 XML Download
XML Download