

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Atherosclerosis and coronary artery disease (CAD), which is developed as a complication of atherosclerosis, are one of the major causes of death in the world (1). Traditional risk factors of coronary artery disease are hypertension, diabetes mellitus, dyslipidemia, and smoking. However, these factors can only explain approximately two thirds of the observed clinical events. This has maintained interest in other nutritional, biochemical, and genetic factors that might contribute to the underlying pathophysiology of vascular disease (2).
Cancers as well as CAD are also the main causes of death in developed countries. They share common disease origin and pathogenetic mechanisms (3). Recent studies addressing the issue of somatically acquired DNA mutations in the pathogenesis of atherosclerosis suggest that the occurrence of DNA alterations contribute to the multifaceted pathogenesis of the atherogenic process. In particular, deletions or mutations of gene coding for enzymes involved in the metabolism of hazardous compounds may be responsible for individual susceptibility to genotoxic factors, predisposing to the development of DNA insults (4, 5). Also twin and nuclear family studies provide compelling evidence of a genetic component underlying cardiovascular disease (6).
The glutathione S-transferases (GST) are a family of enzymes that detoxify reactive electrophiles, particularly in tobacco smoke, products of oxidative stress, and known or suspected carcinogenic compounds such as benzo[a]pyrene and other polycyclic aromatic hydrocarbons (7). There are four classes of GST isoenzymes in humans (µ, π, θ, and α), with partially overlapping substrate specificities (8). GST M1 enzyme belongs to the µ class and the GSTT1 enzyme to the θ class. The GSTM1 gene on chromosome 1p13, according to the three alleles, can be grouped into two classes: GSTM1-null homozygote for the null allele (GSTM1-0), nonfunctional class and GSTM1-1 with at least one of the GSTM1a or GSTM1b alleles, functional class (9). GSTM1-null was reported to be associated in some studies with increased susceptibility to inflammatory pathologies and increased risk of smoking-related cancers (10, 11).
Similarly, GSTT1 gene on chromosome 22q11,2 has two classes denoted as GSTT1-null homozygote for the null allele (GSTT1-0), and GSTT1-1 with at least one functional allele. The GSTT1 enzyme encoded by GSTT1-1 catalyzes the detoxification of monohalomethanes and ethylene oxide, present in cigarette smoke (12), and deficiency of its activity is associated with high risk of smoking-related cancers (13).
Tobacco smoke is a major cause of both cancer and cardiovascular diseases. Although its carcinogenic role via induction of DNA damage and mutation is well established, the mechanisms involved in vascular disease remain unclear. One possible cause is that DNA damage causes smooth muscle cell proliferation in the intima of arteries, thereby contributing to atherothrombotic processes. The binding of chemicals to DNA is modulated by detoxification enzymes (14). Mutagenic and mitogenic activity of cigarette smoke chemicals can cause DNA adducts in target tissues and the oxidative modification of lipoproteins (15), endothelial cell regeneration (16), and progression of atherosclerotic lesions (15). Epidemiological studies have indicated that GSTM1 and GSTT1 polymorphisms are associated with increased risk for different cancers among smokers, but there are still some controversies about the relationship between GST polymorphism and CAD in smokers (7, 17).
The GST enzyme is implicated in the detoxification of carcinogens present in tobacco smoke and consequently polymorphisms in this gene may confer susceptibility to cardiovascular disease if DNA damage is important in the development of CAD. So the aims of this study are to assess whether the GSTM1 and GSTT1 genotypes are associated with CAD, and to ascertain whether the risk of CAD given exposure to cigarette smoking is modified by the specific genetic polymorphisms of GSTM1 and GSTT1.
MATERIALS AND METHODS
Subjects
The study population consisted of 775 patients who had undergone coronary angiography from June 2004 to March 2005 in Kyunghee University Hospital. Most of them (703 of 775) had received the coronary angiography for confirmative diagnosis and treatment of ischemic heart disease with typical chest pain, and 72 subjects for preoperative evaluation of valvular heart disease or other reasons. We classified the subjects into 3 groups according to the degree of luminal diameter stenosis in coronary angiography: group A represents subjects with more than 50% of luminal diameter stenosis, group B with luminal diameter stenosis between 20% and 50%, and group C with less than 20%. Smoking history, hypertension, diabetes mellitus, family history of cardiovascular disease, and obesity were evaluated by history taking and physical examination. Smoker was defined as both current smoker and ex-smoker who had previously been exposed to smoking for at least 5 yr. Hypertension was defined as patients who had been previously diagnosed and received anti-hypertensive drugs, or patients whose blood pressure level was more than 140 mmHg in systole or more than 90 mmHg in diastole. Diabetes mellitus was defined as patients who had been previously diagnosed and received oral hypoglycemic agents or insulin therapy, or patients whose fasting blood glucose (FBS) was above 126 mg/dL or postprandial 2 hour blood glucose (PP2hr glucose) above 200 mg/dL. Dyslipidemia was defined as patients who had taken statin treatment with diagnosis of hyperlipidemia or patients whose low-density lipoprotein cholesterol (LDL) level was above 160 mg/dL or high-density lipoprotein cholesterol (HDL) level below 40 mg/dL. Body mass index (BMI, kg/m2) was calculated using the height and weight. Informed consent was obtained from all subjects, and the study was approved by the Ethical Committee of Kyunghee University.
Methods
Blood test
We determined the baseline blood test including the FBS, PP2hr glucose and lipid profile such as total cholesterol, triglyceride, LDL and HDL.
Coronary angiography
Coronary angiography was carried out according to the Judkins techinique, and images of the right and left coronary trees were obtained in routine standardized projections with Philips Integris Allura 9 system (Philips Medical, Eindhoven, Netherlands). Quantitative coronary angiography was executed by experienced angiographers who were blind to the results of genotype analysis. CAD was defined as luminal diameter narrowing more than 50% in at least one epicardial coronary artery. The severity of CAD was determined by the number of affected vessels in coronary arteris (one-, two-, or three-vessel disease). Also we devided the CAD according to the degree of luminal narrowing into mild (50-75%), moderate (75-90%), and severe (more than 90%).
Genetic assessment
Blood samples and DNA isolation Venous blood of 3 mL was obtained from all subjects and collected in sterile tubes containing ethylenediaminetetraacetic acid (EDTA). Immediately after collection, whole blood was stored at -20℃ until use. Genomic DNA was extracted from whole blood by using Whole Blood Genomic DNA Purification (Rapid Spin Type, CoreBio, Indianapolis, IN, U.S.A.)
Analysis of GSTM1 and GSTT1 gene polymorphisms The genetic polymorphism analyses for the GSTM1 and GSTT1 gene were determined by the multiplex polymerase chain reaction (PCR) modifying the previously described method (18). The appropriate fragment of the GST gene for GSTM1 and GSTT1 was amplified with specific primers from human genomic DNA. The CYP1A1 gene was coamplified as an internal positive control. The following primers were used in PCR reaction: GSTM1 primers of (sense) 5-GAACTCCCTGAAAAGCTAAAGC-3 and (antisense) 5-GTTGGGCTCAAATATACGGTGG-3, GSTT1 primers of (sense) 5-TTCCTTACTGGTCCTCACATCTC-3 and (antisense) 5-TCACGGGATCATGGCCAGCA-3, and CYP1A1 (sense) 5-GAACTGCCACTTCAGCTGTCT-3 and (antisense) 5-CAGCTGCATTTGGAAGTGCTC-3.
PCR was performed in a total volume of 30 µL reaction mix containing 100 µg genomic DNA, 5ρM of each primer, 2.5 mM deoxyribonucleoside triphosphates, 1.5 mM MgCl2, 100 mM Tris-HCL, and 1 U thermostable Taq DNA polymerase with GeneAmp PCR system 2700 (Applied Biosystems, Foster, CA, U.S.A.). The amplification conditions were initial denaturation at 94℃ for 5 min followed by 35 cycles of denaturation at 94℃ for 45 sec, annealing at 59℃ for 50 sec, extension at 72℃ for 1 min, and final extension at 72℃ for 10 min. The products of the PCR amplification (GSTM1:215 bp, GSTT1:480 bp, CYP1A1:312 bp) were separated then electrophoretically on an ethidium bromide-stained 2% agarose gel.
Statistical analysis
Statistical analysis of data was conducted with SPSS 12.0 program. Data of continuous variables were expressed as mean ±standard deviation and data of noncontinuous variables as frequency (N, %). Differences of means among group A, B, and C were evaluated by one-way analysis of variance (ANOVA). Comparison of noncontinuous variables and genotype distribution among 3 groups were tested by using χ2 test (2 × 2 continency table). We calculated odds ratios (OR) and 95% confidence interval (CI) for the association of the GSTM1 and GSTT1 genotypes with the development of CAD using unconditional logistic regression. The OR was also adjusted for other coronary risk factors. Independent factors on CAD risk were evaluated by multiple logistic regression model. p values of <0.05 were considered statistically significant.
RESULTS
Clinical characteristics
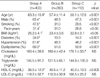
The clinical characteristics for 775 subjects are shown in Table 1. Of 775 subjects, 403 subjects belonged to group A, 260 to group B, and 112 to group C. The prevalence of known atherogenic risk factors, such as smoking habits, hypertension, diabetes, dyslipidemia, obesity, and sex (male) were significantly higher in group A as compared with group B and C. We found that the HDL level was significantly lower in group A than in group B and C (36.0±12.0 in group A vs. 40.5±11.3 in group B, and vs. 40.2±10.5 mg/dL in group C, respectively, p<0.05). Average smoking history was 30.2 pack years. There was no difference in smoking habits between group A and B, but there was significant difference between group A and C (31.4 pack years in group A vs. 26.8 pack years in group C, p<0.05). There was no significant difference in clinical characteristics between group B and C.
GSTM1/T1 gene polymorphism
The internal standard fragment amplified from GSTM1 gene was 215 bp. A 480-bp fragment was amplified for the GSTT1 gene, and 312-bp fragment was obtained for CYP1A1 gene. The absence of other amplified products with only presence of 312-bp fragment was consistent with the null genotypes (Fig. 1).
We acquired data for GSTM1 gene and GSTT1 gene polymorphisms in 692 subjects (89.3%) due to missing data of gene analysis (missing data [83]: group A [47], B [34], and C [2]).
The relation of GSTM1/T1 gene polymorphism to coronary artery disease
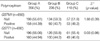
There was no significant difference in GSTM1 and GSTT1 genotype distribution among 3 groups (Table 2). We found no difference in genotype distribution among 3 groups when we classified subjects into smokers and non-smokers. When the risks associated with GST genotypes for CAD were considered in relation to smoking habits, smokers with GSTM1 null genotype were at an approximately 2.07-fold higher risk of CAD than nonsmokers with GSTM1-positive genotype (CI, 1.06-4.07, p=0.03). However, smokers with GSTM1-positive genotype had no increased risk for CAD as compared with non-smokers (OR, 1.21, CI, 0.64-2.27). In non-smokers, there was no difference in CAD risk according to GSTM1 genotype (OR, 0.96, CI, 0.56-1.66). There was no effect of interaction of GSTM1 genotype and smoking on CAD development between group B and C (Table 3).
The effect of interaction of GSTT1 gene and smoking on CAD risk, we found the similar results as those in GSTM1 gene. Smokers with GSTT1 null genotype were at an approximately 2.0-fold higher risk of CAD than nonsmokers with GSTT1 positive genotype (CI, 1.05-3.84, p=0.03). However, smokers with GSTT1-positive genotype had no increased risk for CAD as compared with non-smokers (OR, 1.44, CI, 0.74-2.84). In non-smokers, there was no difference in CAD risk in relation to GSTT1 genotype (OR, 0.77, CI, 0.45-1.31). There was no effect of interaction of GSTT1 genotype and smoking on CAD development between group B and C(Table 4).
Considering the synergistic effects of GSTM1 null genotype and GSTT1 null genotype on risk of smoking-related CAD, we found that smokers with both GSTM1 and GSTT1 null genotype were at an approximately 2.76-fold higher risk of CAD than nonsmokers with both GSTM1 and GSTT1 positive genotype (CI, 1.17-6.52, p=0.04)
After adjusting other coronary risk factors such as hypertension, diabetes, dyslipidemia, age, and sex than smoking, the interactive effect GSTM1/GSTT1 gene and smoking on CAD remained statistically significant (in smokers with GSTM1 null genotype, OR, 2.01, CI, 1.13-3.69, p=0.01/in smokers with GSTT1 null genotype, OR, 1.89, CI, 1.03-3.13, p=0.02) (Table 5, 6).
As we considered the effect of GSTM1/GSTT1 genotype on the multivessel disease, there was no significant difference in GSTM1/GSTT1 genotype in relation to the number of involved vessel in both smokers and non-smokers (Table 7). Also, there was no significant effect of GSTM1/GSTT1 genotype on the severity of CAD (Table 8).
We examined independent risk factors for CAD by multiple logistic regression analysis. Smoking, hypertension, diabetes, old age more than 50 yr, male, and dyslipidemia were independent risk factors for CAD. Both GSTM1 and GSTT1 null genotype in smokers were also independent risk factors, but GSTM1/T1 null genotype itself was not an independent risk factor (Table 9).
DISCUSSION
Cardiovascular diseases and cancer are the main causes of death in developed countries to which cigarette smoking is a major contributing factor. Cancer and atherosclerosis share common risk factors, such as cigarette smoking, physical inactivity, exposure to certain environmental mutagenic agents, and dietary habits. Autopsy studies have demonstrated higher risk for severe atherosclerosis in persons who died from tobacco-related cancers than those who died from extraneous causes (19). The facts that monoclonal proliferation of vascular smooth muscle cells in atherosclerosis is similar to process in carcinogenic progression (20) and that somatic mutation contributes to the developement of atherosclerosis (4) suggest that cardiovascular diseases and cancers share common pathogenic mechanisms.
Epidemiological studies have demonstrated tobacco smoke is a major cause of both cancer and vascular diseases. However, the mechanism by which exposure leads to disease is better understood in the former case. There are many identified carcinogens in tobacco smoke that induce DNA damage by direct binding to form DNA adducts (21). Unrepaired DNA damage can result in the induction of somatic mutations in genes regulating cell growth, thus providing a mechanism for carcinogenesis.
By contrast, the mechanism by which smoking contributes the development of vascular diseases is not certain yet. One possible explanation is that smoking-induced DNA damage causes smooth muscle cell proliferation in the intima of arteries, thereby contributing to atherothrombotic processes (14). The hypothesis that DNA damage plays a role in vascular disease has received support from observations in animal models and humans. In experimental animals, chemicals in tobacco smoke (e.g., benzo(a)pyrene, 1,3-butadiene) and environmental tobacco smoke have been reported to induce and stimulate atherosclerotic plaque formation (11). In addition, in both humans and animals, tobacco carcinogens induce DNA adducts in cells of the vasculature at high levels, and aromatic DNA adducts are present in smooth muscle cells of human atherosclerotic lesion of the abdominal aorta and in heart tissue (22).
GST is a family of enzymes that detoxify reactive electrophiles, particularly present in tobacco smoke, reactive oxygen species, and known or suspected carcinogenic compounds (7). Modulation of DNA damage and mutation caused by polymorphisms in detoxification enzymes, including the GST, is a well-established risk factor for tobacco-related carcinogenesis and a similar change in cellular damage may be involved in the risk of vascular disease associated with tobacco smoking (23). Also the binding of xenotoxic chemicals to DNA is modulated by detoxification enzymes such as GST. Of four classes of GST isoenzymes, null genotypes of GSTM1 which belongs to the µ class and of GSTT1 which is in the class are associated with increased risk of smoking-related cancer (10, 11, 13). However, association of GSTM1/T1 gene polymorphism with smoking-related CAD is revealed in few studies and the results of these studies are different (7, 17, 24).
In this study, we tested whether specific genotypes of GSTM1 or GSTT1 may affect susceptibility to smoking-related CAD. The null genotype of GSTM1 and GSTT1 in smokers were associated with an increase in the risk of CAD (OR, 2.07, CI, 1.06-4.07, p=0.03; OR, 2.0, CI, 1.05-3.84, p=0.03), respectively. The effects of GSTM1/GSTT1 genotype on CAD were augmented when both GSTM1 and GSTT1 genotype were null type (OR, 2.76, CI, 1.17-6.52, p=0.04). However, smokers with GSTM1/GSTT1-positive genotype had no increased risk for CAD as compared with non-smokers (OR, 1.21, CI, 0.64-2.27; OR, 1.44, CI, 0.74-2.84, respectively). In non-smokers, there was no difference in risk for CAD in relation to GSTM1/GSTT1 genotype (OR, 0.96, CI, 0.56-1.66; OR, 0.77, CI, 0.45-1.31, respectively). These facts suggest that there may be different disease susceptibility to CAD according to the GSTM1/GSTT1 genotypes in smokers.
As mentioned above, there are still some controversies about the relationship between GST polymorphism and CAD in smokers. An epidemiological study showed that smokers with GSTM1 null genotype have a consistently higher prevalence of CAD (7). De Waart and colleagues reported that the 2-yr progression of common carotid intima media thickness was clearly more increased in smokers with the GSTM1 null genotype than in smokers with the GSTM1-positive genotype (25). Recent data also suggest that the GSTM1 null genotype predisposes subjects to severe CAD (26). Our study showed similar results to those previously reported. By contrast, another study reported a significantly decreased risk for acute myocardial infarction in smoking patients with GSTM1 null genotype, but only in those having a previous history of the same disease (17).
Meanwhile, previous studies showed the more diverse results about the effects of GSTT1 gene polymorphism on smoking-related CAD. A recent study and our study found that smokers with GSTT1 null genotype had increased risk for CAD (24), but other studies revealed that GSTT1 positive genotype was associated with increased risk for CAD and peripheral artery diseases (7). A possible explanation for these conflicting results is that GSTT1 protein activates some chemicals present in cigarette smoke in more toxic forms and increase the risk of atherosclerosis (7). Indeed, although mammalian GST θ behaves as a scavenger towards electrophiles such as epoxides, it acts also as metabolic activator, producing intermediates potentially dangerous for DNA and cells (27). However, cytogenetic studies have shown that blood cultures from individuals with GSTT1 null and GSTM1 null genotypes have increased in vitro sensitivity to various genotoxins resulting in an increased level of chromosomal and oxidative DNA damage in lymphocytes (28). And levels of adducts to DNA in smooth muscle cells from atherosclerotic lesions are consistently increased in individuals having the GSTM1 null genotype (22). Consequently, genetic polymorphisms that affect xenobiotic metabolism or cellular response to DNA damage by modulating individual sensitivity to genotoxins may be important in modulating susceptibility to both cancer and atherosclerosis. In particular, the polymorphic genes of GST family of enzymes, which are involved in the detoxification of many compounds in cigarette smoke, may be relevant to the susceptibility to smoking-related diseases.
The effect of cigarette smoking on atherosclerosis involves many metabolic and biological processes, such as endothelial injury, oxidation of low-density lipoprotein and changes in the hemostatic system (29). In addition, some of the adverse effects of smoking may result from DNA damage by genotoxic and carcinogenic agents including polycyclic aromatic hydrocarbons, n-nitrosamines, and reactive oxygen species in cigarette smoke.
Our data may provide additional information to the understanding of smoking-related cardiovascular risk. Although there are several mechanisms of smoking-related CAD, it is thought to be important that deleted polymorphisms in the GST genes may influence the susceptibility to smoking-induced CAD by modulating the detoxification of genotoxic atherogens.








 XML Download
XML Download