

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pelizaeus-Merzbacher disease (PMD) is a rare, X-linked recessive dysmyelinating leukodystrophy of the central nervous system (CNS). It is caused by mutations in the proteolipid protein 1 (PLP1) gene on the long arm of the X chromosome in band Xq22 that encodes a major component of CNS myelin protein (1, 2). There are several related tests available for the diagnosis of PMD; however, no definitive test exists. In spite of this, molecular testing of PLP1 is now essential to confirm diagnosis. Here, we present a Korean boy with PMD who had clinical symptoms mimicking cerebral palsy and was confirmed to have a PLP1 duplication mutation using a specific molecular technique.
CASE REPORT
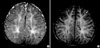
A 52-month-old boy with developmental delay was admitted to the department of rehabilitation medicine for evaluation and treatment. He was born post-maturely at the gestational age of 41 weeks with an initial body weight of 3,400g. The mother underwent a cesarean section due to failure of progression during labor; however, the pregnancy and prenatal period of the patient had been uneventful. The boy showed significantly delayed development. He could turn over and pull-to-stand, but he could not stand without support. Also, genu recurvatum was observed while standing. He could not walk nor keep balance in a sitting position. His kneeling and crawling were inadequate. He could not speak any meaningful word. Physical examination showed horizontal nystagmus, saccadic gaze, intentional tremor, head titubation, and ataxia. The deep tendon reflexes were hyperactive in all extremities and positive ankle clonus was observed, bilaterally. Electromyography was normal but somatosensory evoked potential showed no response on median nerve stimulation. Creatine phosphokinase was normal and lactic dehydrogenase was slightly increased (525 IU/L). Thyroid function tests, liver function tests, and infant hereditary metabolic screening tests (very-long-chain fatty acid, lactic acid, pyruvic acid, taurine acid, urine organic acid, mucopolysaccharide, urine and serum amino acids) were also normal. The brain magnetic resonance image (MRI) studies revealed diffuse abnormal high signal intensities in the white matter tract including observation of subcortical U fiber on the T2-weighted and fluid attenuated inversion recovery (FLAIR) image (Fig. 1). The pattern of the demyelination, which manifested as high SI on T2-weighted or FLAIR images, was patchy, with characteristic sparing of perivascular white matter creating a "tigroid" or "leopard-skin" pattern. There was no abnormal enhancement found during a gadolinium-enhanced study. The overlying cortex was normal.
His younger brother, a 27-month-old boy at present with developmental delay, visited our outpatient clinic. The boy revealed also significant developmental delay. He could creep, but could not sit without support and, could not pull to sit. He showed poor sitting balance while in the sitting position. On physical examination, he also showed horizontal nystagmus, saccadic gaze, rhythmic tremor, and head titubation. The deep tendon reflexes were brisk in all extremities and positive ankle clonus was observed, bilaterally. He was also unable to say any meaningful words.
After obtaining informed consent, blood samples were collected from the patient, his parents and younger brother for a genetic study. The genomic DNA was isolated from peripheral blood leukocytes using a Wizard genomic DNA purification kit according to the manufacturer's instructions (Promega, Madison, WI, U.S.A.). We used multiple ligation-dependent probe amplification (MLPA) to screen all exons of the PLP1 gene using a MLPA kit (SALSA P022; MRC Holland, Amsterdam, Netherlands). The kit contains a total of 32 probes, including 1 probe for each of the 7 PLP1 exons, 8 control probes of other parts of the X chromosome, 1 probe that detects a Y-chromosome, and 16 probes that detect sequences on autosomes. All thermal reactions were carried out on a thermal cycler (Model 9700, Applied Biosystems, Foster City, CA, U.S.A.). One hundred nanograms of sample DNA were diluted to 5 µL with deionized water and denaturated at 98℃ for 5 min before the addition of 3 µL MLPA probe mix and buffer. The reaction mixture was denaturated for 1 min, then incubated for 16 hr at 60℃ to ensure specific hybridization of the oligonucleotide probes with their target sequences. After hybridization, a buffer/ligase mixture (32 µL) was added according to the manufacturer's instructions and incubated at 54℃ for another 15 min to stabilize the ligation reaction. Ligation was terminated by heating to 98℃ for 5 min. A fluorescent multiplex polymerase chain reaction (PCR) amplification using a single universal primer pair suitable for all 40 probes in each kit was carried out under standard conditions (annealing temperature 60℃, 36 cycles). PCR products were analyzed on a fluorescent capillary sequencer (ABI 3100, Applied Biosystems, Foster City, CA, U.S.A.) using Genescan software. Five healthy female control samples were included in each MLPA test to calibrate unknown samples. Data analysis was performed with GeneMarker® software (SoftGenetics LLC, State College, PA 16803, U.S.A.). The PLP1 gene dosage was determined using a population normalization method. Expected PLP1 values for non-PMD males, non-PMD females, males with a PLP1 duplication, and female carriers of a PLP1 duplication are 0.5, 1.0, 1.0 and 1.5, respectively.
We determined the relative PLP1 copy numbers from peak areas by normalizing each PLP1 probe to each autosome, using normal female DNA as a calibrator. The patient and younger brother had all 7 PLP1 exons duplicated. His father showed normal relative copy numbers of the PLP1 gene but his mother showed findings consistent with carriers of a duplication. The relative dosage of PLP1 exons in the patient and his family members are summurized in (Fig. 2). Taken together, these findings indicate the patient and his younger brother could be diagnosed with PMD caused by duplicated PLP1 gene, inherited from their carrier mother.
DISCUSSION
Clinical findings in cases of PMD are very different and few studies have been reported. Golomb et al. conducted a study of 10 boys and reported their clinical findings (3). They found that prenatal complication occurred in eight patients and that their patients demonstrated defect of social interaction, dysphasia, nystagmus, hypotonia, and spasticity, that affected the legs more than the arm, as well as ataxia. The patient in our study demonstrated horizontal nystagmus, saccadic gaze, intentional tremor, head titubation, and ataxia. The deep tendon reflexes were hyperactive in all extremities. Unlike general cases, the leg and arm were equally involved and frequently showed spastic quadriplegia as seen in cases of cerebral palsy, and initially, our clinics suspected that the boy had spastic quadriplegia due to cerebral palsy. We focused on clinical diagnosis such as spasticity, inability to walk, and MRI findings around the periventricular white matter. The patient in our study showed only mild spasticity and genu recurvaturm. Due to mild spasticity, the patient did not show equines of foot or contracture of hip adductors, which have been frequently shown in spastic quadriplegic type cerebral palsy.
PMD has been reported several times in Korea since 1995, but they were diagnosed by the clinical features and MRI findings (4-6). In 2004, the first Korean case of PMD with a confirmed point mutation in PLP1 was reported (7); however, aside from this case, there have been no reports of confirmation based on a genetic study.
Barkovich (8) suggested that in PMD, MR imaging showed reduction of normal myelin and normal oligodendroglial cells in the brain, resulting in delayed myelination. Due to the marked reduction of myelin, both diffusion anisotropy and magnetization transfer were decreased compared to age-matched control subjects. Our case showed abnormally high signal intensities in the white matter tract, including subcortical new fiber on the T2-weighted and FLAIR image, which indicates abnormal myelination of white matter in the brain.
Complete duplication of the PLP1 gene on Xq22 is the cause of 60-70% of PMD cases, whereas deletions of this gene as well as point mutations in coding or splice site regions are involved in most of the remaining cases. Duplications of PLP, indicating an increased PLP1 dosage, prevent them from making normal myelination. Patients with an increased PLP1 dosage have variable phenotype ranging from severe connatal to mild PMD, but most have a classic form of disease. Carrier females with a PLP1 duplication are usually asymptomatic (9).
Various methods have been used to identify PLP1 gene duplications, including fluorescent in situ hybridization (FISH) (10), array comparativ genomic hybridization (CGH) (11), quantitative PCR (12), MLPA (13), real-time PCR (14), and multiplex amplification and probe hybridization (MAPH) (15). MLPA is based on the ligation of 2 adjacent annealing oligonucleotides followed by quantitative PCR amplification of the ligated products. Previous studies have confirmed the efficiency of MLPA for detecting PLP1 gene duplications and deletions with good intra- and interassay precision (16). The precision of MLPA analysis was excellent and comparable to or better than quantitative real-time PCR, with CVs of 4.3-9.8% and 3.0-14%, respectively. Furthermore, MLPA analysis using customized probe sets has proven to have significant advantages over other methods such as FISH or array-CGH or MAPH as a rapid, reliable, economical, and easy molecular test (17-19).
In summary, this is the first reported Korean case of PMD in two affected male siblings with a confirmed duplication of the PLP1 gene, inherited from their mother, an asymptomatic carrier. Family study of the PLP1 gene in Korean patients with suspected PMD, therefore, should be considered to better understand the mechanism of genetic disorders as well as for counseling and family planning.
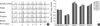
 XML Download
XML Download