

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Fibrosis has been known to be the main pathogenesis in the progression or severity of various lung diseases including chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF). According to one recent study by Hogg, it is the fibrosis of small airways that is associated with the severity of COPD (1). Also, the recent paradigm suggests that IPF is the result of epithelial injury and subsequent deregulated repair resulting in excess extracellular matrix formation and abnormal mesenchymal cell activation and proliferation (2). While resident fibroblast in injury area is traditionally considered to be the principal mediator of fibrosis, recent studies have highlighted the involvement of epithelial cells via an epithelial to mesenchymal transition (EMT) during tissue repair following injury (3-5). Transforming growth factor β1 (TGF-β1) has been identified as the main inducer of EMT in kidney and lung diseases including IPF and COPD (7, 8). During EMT, the intercellular adhesion molecule E-cadherin appears to have a central role, since the loss of E-cadherin expression correlates with the ability of epithelial cells to adopt mesenchymal phenotypes. While there are various lung diseases of which fibrosis is the main pathogenesis, few studies have been conducted on the EMT in lung diseases (9, 10). To understand the role of EMT in lung fibrogenesis, we examined whether TGF-β1 can induce EMT in A549 cells and the concentration of TGF-β1 to induce A549 cells to undergo EMT. IL-1β has been suggested to induce kidney epithelial cells to undergo EMT (11). We examined whether IL-1β alone had effects to induce A549 cells to mesenchymal-like cells transition. Recently, a transcription factor called snail has been described as a potent repressor of E-cadherin expression and an inducer of epithelial to mesenchymal transition. To determine whether snail expression was associated with the phenotypic changes of A549 cells, we used reverse transcriptase-polymerase chain rection (RT-PCR) analysis.
MATERIALS AND METHODS
Materials
Recombinant human TGF-β1 and human IL-1β were purchased from R&D systems. Monoclonal antibodies against E-cadherin (R&D systems, Minneapolis, MN, U.S.A.), pancytokeratin (Sigma Aldrich, Saint Louis, MO, U.S.A.), vimentin (Sigma Aldrich, Saint Louis, U.S.A.), and rhodamine-phalloidin (Molecular probes, Inc., Eugene, OR, U.S.A.) were used. Fluorescent-conjugated secondary antibodies labeled with goat anti-mouse IgG (Alexa Fluor 488; Molecular probes, Inc., Eugene, OR, Califonia, U.S.A.) were used. The fluorescent images were obtained by confocal laser scanning microscope (Zeiss LSM 510, Oberkochen, Germany).
Cell culture
Human type II alveolar epithelial cells (A549) were supplied from KCLB (Seoul, KOREA). Cells were maintained in L-glutamine-RPMI 1640 containing 10% FBS, 25 mM HEPES, 100 U/mL penicillin and 100 µg/mL streptomycin at 37℃ in a humidified 5% CO2 atmosphere. Confluent cultures of cells were maintained in serum-free RPMI containing 0.1% FBS for 24 hr prior to stimulation with cytokines. The cells were incubated with various concentrations of the cytokines for 48 hr. For immunofluorescence studies, cells were seeded on 22 mm round cover slips coating collagen type I (BD biosciences, Bedford, MA, U.S.A.). The morphologic features of cells in confluent cultures were shown to remain stable during the two cell passages. After each passage, the cells grew to confluence within 4 to 5 days.
SDS-PAGE and Western blot
Cells were washed twice with ice-cold PBS, lysed with lysis buffer (50 mM Tirs-Cl pH7.5, 150 mM NaCl, 2 mM EDTA, 1% NP-40) on ice for 30 min, and scrapped into a centrifuge tube. The cell lysates were centrifuged at 13,000 rpm for 20 min and were measured by the method of the Bradford solution (Bio-rad, CA, U.S.A.). Total cellular protein was mixed with SDS sample buffer and reducing agent β-mercaptoethanol and heated at 95℃ for 5 min. The samples were then subjected to electrophoresis in 10% SDS-PAGE gel and transferred to Hybond ECL membrane. After the transfer, the blotting membranes were incubated for 2 hr at room temperature in TBST (TBS containing 0.1% [v/v] Tween 20) with 5% (w/v) skim milk to block nonspecific binding sites. Then, the blot was incubated for 2 hr in blocking solution with primary antibody at room temperature (RT). After washing the blot with TBST, it was incubated with a horseradish peroxidase-conjugated secondary antibody. After extensive washing, the immunoblots were visualized by ECL kit (Amersham Life Science, Inc., Piscataway, NJ, U.S.A.).
RT-PCR
Total RNA was isolated using Trizol (Invitrogen, Califonia, U.S.A.). The complementary DNA was obtained from 500 ng-1 µg of total RNA with the use of the Superscript™ III First-Strand Synthesis system and Platinum PCR SuperMix (Invitrogen Carlsbad, CA, U.S.A.). Amplification of snail was performed for 40 cycles (40 sec at 95℃, 30 sec at 53℃, and 1 min at 72℃) with the use of primer 1 (5'-CACATCCTTCTCACTGCCATG-3') and primer 2 (5'-GCATCTAAACTCTAGTCTGC-3'). For nested RT-PCR analysis of snail, a 30-cycle reaction was performed under the same condition, and a 1:50 dilution of the product of the reaction was amplified for 20 cycles (40 sec at 95℃, 30 sec at 60℃, and 1 min at 70℃) with primer 1 and primer 3 (5'-CCTGAGTGGGGTGGGAGCTTCC-3'). PCR analysis of E-cadherin was performed for 32 cycles (30 sec at 94℃, 30 sec at 60℃, and 1 min 72℃). GAPDH were amplified using 25 cycles (initial denaturizing at 94℃ 2 min followed by PCR amplification, 94℃/min, 61℃/min, and 72℃/min) with primer 1 (5'-GCTCTCCAGAACATCATCCCTGCC-3') and primer 2 (5'-CGTTGTCATACCAGGAAATGAGCTT-3').
Immunofluorescence studies
Cells were cultured on 22-mm round cover slip (BD biosciences, Bedford, MA, U.S.A.) until confluence developed, and then were treated with TGF-β1 at 0.5-5ng/mL, IL-1β at 2 ng/mL for 48 hr. The level of concentration of IL-1β used for this study was determined based on studies showing IL-1β induced EMT and the lowest level of concentration of TGF-β1 to induce EMT was selected to see mainly IL-1β induced effects. The cells were washed, fixed in ice-cold methanol or 4% paraformaldehyde for 10 min, and then soaked in PBS containing 0.1% Triton X-100 for 10 min to increase their permeability to antibodies. After washing the cells three times for 5 min in PBS, they were blocked with 5% BSA in PBS for 3 hr at RT. The incubation with primary antibody was performed for 2 hr at RT. Bounded primary antibodies were detected using FITC-conjugated secondary antibodies. The fluorescent images were obtained by confocal laser scanning microscope (Zeiss LSM 510).
RESULTS
The concentration of TGF-β1 to induce EMT in A549 cells
To demonstrate epithelial-to-mesenchymal transition in vitro, we first examined TGF-β1 concentrations to induce EMT in cultures of alveolar type II epithelial cell line, A549. Changes in cell morphology were also assessed under phase contrast light microscopy (Fig. 1A), and the expression of E-cadherin and cytokeratin, the epithelial phenotype markers, and of F-actin and vimentin, mesenchymal phenotype markers, were determined following treatment of A549 cells with various TGF-β1 concentrations (0.05-5 ng/mL) for 48 hr (Fig. 1B). The assessment under phase contrast light microscopy demonstrated that cells began morphologic changes at the 0.05 ng/mL TGF-β1 concentration (Fig. 1A) and changed to more fibroblast-like cells in a concentration-dependent manner (Fig. 1B). The loss of E-cadherin expression was revealed at TGF-β1 concentration levels as low as 0.05 ng/mL and in a concentration-dependent manner. E-cadherin expression at 0.5 ng/mL TGF-β1 concentration totally disappeared. However, cytokeratin suppression was not as profound as that of E-cadherin (Fig. 1B).
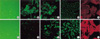
Epithelial-to-mesenchymal transition of A549 cells in vitro
A549 cells cultured in the absence of TGF-β1 maintained the classic cobblestone epithelial morphology and growth pattern, but after treatment of 5 ng/mL TGF-β1 concentration for 48 hr, the cells adopted a more fibroblast-like morphology and showed a reduced cell-cell contact. Confocal immunofluorescence microscopy demonstrated the loss of E-cadherin and the reorganization of the actin cytoskeleton from the cortical band typical of epithelial cells to fibroblastic stress fibers after TGF-β1 treatment. Also, cytokeratin was replaced by vimentin, although some fibroblast-like cells were still positive for cytokeratin (Fig. 2).
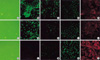
Effects of IL-1β alone or in combination with TGF-β1 on EMT
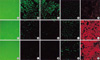
A549 cells were treated with either IL-1β or TGF-β1 alone or with IL-1β in combination with TGF-β1. IL-1β alone demonstrated more elongated cells morphology under phase contrast light microscopy and weaker E-cadherin and cytokeratin expression than in the control (Fig. 3), but stress fiber reorganization by F-actin was not observed. The combination of IL-1β with TGF-β1 induced A549 cells to more fibroblast-like cells (Fig. 3). Another study using TGF-β1 at a low concentration (0.5 ng/mL) also demonstrated down-regulation of E-cadherin and cytokeratin expression and stress fiber reorganization by F-actin. The expression of mesenchymal phenotypic markers was more profound with the addition of IL-1β to TGF-β1. There was more cytokeratin replacement by vimentin and stress fiber reorganization by F-actin (Fig. 4). Our data suggested that IL-1β alone was not enough to induce EMT but showed synergic effects with TGF-β1 in the EMT phenomenon.
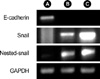
TGF-β1 induced EMT is accompanied by snail expression
While no snail messenger RNA (mRNA) signals were found without TGF-β1 treatment, expression of snail mRNA was revealed after TGF-β1 treatment. Stimulation of A549 cells with TGF-β1 plus IL-1β revealed increased snail expression (Fig. 5). Our data suggested that EMT in A549 cells might be associated with snail expression and IL-1β might have synergistic effects in snail expression.
DISCUSSION
EMT is necessary for embryonic development and tumor progression (12, 13). In recent years, EMT in adult is speculated to occur involving resident epithelia in response to injury, as an additional source of myofibroblasts/fibroblasts, which are essential for repair of injured tissue (14, 15). Numerous independent studies have demonstrated that FSP1 (S1004A), a member of the S100 family of calcium-binding proteins exclusively expressed in fibroblasts, can be detected in tubular cells of injured kidneys in the process of EMT in different animal models of chronic renal disease and also in human kidney biopsies (16-18). One study utilizing a model of γGT-LacZ transgenic mice supported evidences that more than one third of renal interstitial fibroblasts were derived from renal tubular epithelium via EMT (14). TGF-β1 is a potent inducer of extracellular matrix formation and has been implicated as the key mediator of lung fibrogenesis. Thus, one possible role of TGF-β1 in lung diseases is to induce alveolar epithelial cells to undergo EMT. Our present data support that TGF-β1 exposure of A549 cells induced EMT characterized by loss of epithelial marker E-cadherin, cytokeratin replacement by vimentin, transformation of myofibroblastic morphology, and stress fiber reorganization by F-actin. A study to detect the concentration of TGF-β1 in loss of E-cadherin using western blotting demonstrated that E-cadherin began to disappear at the concentration of 0.05 ng/mL TGF-β1 in a concentration-dependent manner. Although IL-1β has been known to induce kidney epithelial cells to undergo EMT, some studies have shown that IL-1β failed to induce other cell types to undergo EMT (19). We therefore tested whether IL-1β had similar properties to TGF-β1 in inducing A549 cells to form mesenchymal-like cells and an additive morphologic effect of both stimuli was observed. The lowest concentration of TGF-β1 inducing EMT was used to see mainly IL-1β effects. Cells changed to elongated cell morphology with IL-1β treatment under phase light microscopy. IL-1β alone decreased the expression of E-cadherin and cytokeratin, but the expression of mesenchymal markers vimentin and stress fiber reorganization was not observed. The combination of IL-1β with TGF-β1 resulted in weaker E-cadherin expressions and cytokeratin replacement by vimentin, and increased stress fiber reorganization by F-actin than with TGF-β1 alone. The increased snail expression resulting from the stimuli of TGF-β1 plus IL-1β supports the synergic effect of IL-1β. Our results show that the IL-1β failed to induce A549 cells to undergo EMT, possibly due to the differences in the cells types investigated; however, additive morphologic effects were revealed under IL-1β in combination with TGF-β1. Our data are too limited to clearly demonstrate the pathogenesis of IL-1β-induced synergy effects on EMT, but there is a possibility that IL-1β might function through a TGF-β1-dependent mechanism when we considered other studies of IL-1β induced EMT. While numerous distinct signaling pathways have been described as initiators of EMT in different settings, all of them culminate in the loss of E-cadherin (20, 21). Several studies have demonstrated that E-cadherin is an important determinant for the maintenance of the epithelial phenotype (22, 23). Recent results demonstrated that suppression of E-cadherin expression alone, by the transcription factor snail, induced EMT in carcinoma cells (24). Also, different studies demonstrated that loss of cell-cell adhesion is due to decreased E-cadherin expression (25). Repression of E-cadherin may free up more cytoplasmic β-catenin, which is coimported with lymphoid enhancer factor to the nucleus where its activation is strongly associated with EMT (26). Our data showed that EMT process in A549 cells is closely associated with snail expression despite the difficulty to reveal the pathways of snail in loss of E-cadherin. Further studies are required to reveal the pathway of EMT and to investigate whether EMT is truly involved in lung fibrogenesis.

 XML Download
XML Download