

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Peroxisome proliferator-activated receptors (PPARs) are members of the ligand-activated nuclear hormone receptor superfamily (1). Three PPAR isoforms (α, γ and δ) differ in their tissue distribution and ligand specificity (1). PPAR-α is present in heart, kidney, and liver and appears to primarily regulate genes involved in lipid and lipoprotein metabolism (1, 2), whereas PPAR-δ expression is ubiquitous (3, 4) and its physiological role is unclear (5). PPAR-γ is expressed predominantly in adipose tissue, with lower levels in muscle and liver (6, 7). Although the physiological role of PPAR-γ has not been clearly determined, it appears to be an important transcriptional regulator of genes involved in glucose and lipid metabolism. It was originally characterized as a regulator of adipocyte differentiation and lipid metabolism. PPAR-γ was also shown to be expressed in other cell types, including T lympocytes and chondrocytes (8-10).
PPAR-γ can heterodimerize with at least one other member of the steroid receptor superfamily, retinoid acid receptor (RXR) (11). Ligand-activation of the PPAR-γ:RXR heterodimer has been shown to modulate the transcription of genes involved in lipid metabolism, inflammation, and cell differentiation (12). Recent evidence also shows that the natural receptor ligand for PPAR-γ, 15d-PGJ2 (13), and synthetic antidiabetic thiazolidinedione drugs (e.g. BRL49653, rosiglitazone [Rtz] and ciglitizone), inhibit macrophage and monocyte activation (14) and suppress tumor cell growth (15). Specific ligands for the PPAR-γ and RXR have been shown to act synergistically to induce terminal differentiation of human liposarcoma cells (16) in vitro and to enhance insulin sensitivity in diabetic animals (17).
To maintain cartilage homeostasis, articular chondrocytes synthesize cartilage-specific extracellular matrix molecules, including type II collagen and sulfated proteoglycans (18). This homeostasis is lost in cartilage diseases such as rheumatoid arthritis (RA) and osteoarthritis (OA), eventually leading to cartilage destruction. Cartilage destruction involves a loss of differentiated chondrocyte phenotypes (i.e. dedifferentiation), which is characterized by the cessation of type II collagen expression and onset of fibroblastic type I and type III collagen expression (19). Interleukin-1β (IL-β) and tumor necrosis factor α (TNF α) have been shown to inhibit the synthesis of cartilage components and to promote their degradation by activating metalloproteinases (20). Concurrently to these effects, they induce the production by chondrocytes of inflammatory mediators such as prostaglandins, nitric oxide (NO), and other reactive oxygen species (21). To date most of the drugs marketed as nonsteroidal anti-inflammatory drugs or corticosteroids are unable to prevent cartilage damage (22). Thus, intense investigations are carried out to precise the transduction pathways that impair cartilage homeostasis in order to find new strategies that would be helpful to prevent cartilage destruction.
In this study, we investigated whether 15d-PGJ2, PPAR-γ activator, may modulate the differentiation and inflammation in rabbit articular chondrocytes. We report here that PPAR-γ activation induces dedifferentiation but not inflammation in articular chondrocytes. These results suggest that PPAR-γ agonists may provide a new therapeutic approach for RA and OA.
MATERIALS AND METHODS
Monolayer culture of rabbit articular chondrocytes and experimental culture condition
Rabbit articular chondrocytes were released from cartilage slices of 2-week-old New Zealand white rabbits by enzymatic digestion. Briefly, cartilage slices were aseptically dissected and then dissociated enzymatically for 6 hr in 0.2% collagenase type II (381 U/mg solid, Sigma Louis, MO, U.S.A.) in phosphate-buffered saline, and then individual cells were obtained by collecting the supernatant after brief centrifugation. The cells were resuspended in Dulbecco's modified Eagle's medium (DMEM, Gibco-BRL, Gaithersburg, MD, U.S.A.) supplemented with 10% (v/v) fetal bovine-calf serum, 50 µg/mL streptomycin, and 50 units/mL penicillin, after which they were plated on culture dishes at a density of 5×104 cells/cm2. The medium was changed every 2 days after seeding, and cells reached confluence in approximately 5 days. Differentiation status of articular chondrocytes was determined by examining the accumulation of sulfated glycosaminoglycan with Alcian blue staining or expression of type II collagen (Chemicon, Temecula, CA, U.S.A.) by immunoblot analysis as described previously (23).
Immunoblot analysis
Whole cell lysates were prepared by extracting proteins using a buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, and 0.1% sodium dodecylsulfate, supplemented with protease inhibitors (10 µg/mL leupeptin, 10 µg/mL pepstatin A, 10 µg/mL aprotinin and 1 mM of 4-[2-aminoethyl] benzenesulfonyl fluoride) and phosphatase inhibitors (1 mM NaF and 1 mM Na3VO4). The proteins were size-fractionated by SDS-polyacrylamide gel electrophoresis and transferred to a nitrocellulose membrane. The nitrocellulose sheet was then blocked with 3% non-fat dry milk in Tris-buffered saline. COX-2 was detected using antibody purchased from Cayman Chemical (Ann Arbor, MI, U.S.A.). SOX-9, ERK-2, and PPAR-γ were detected using antibodies purchased from Santa Cruz Biotech (Santa Cruz, CA, U.S.A.). The bands were visualized using peroxidase-conjugated secondary antibodies and chemiluminescence.
Immunohistochemistry
Rabbit joint cartilage explants or arthritic cartilage were fixed in 4% paraformaldehyde in PBS for 24 hr at 4℃, washed with PBS, dehydrated with graded ethanol, embedded in paraffin, and sectioned at 4-µm thickness. The sections were stained by standard procedures using Alcian blue or antibody against type II collagen or COX-2 and visualized by developing with a kit purchased from DAKO (Carpinteria, CA, U.S.A.), following the procedure recommended by the manufacturer.
PGE2 assay
PGE2 production was determined by measuring the levels of cellular and secreted PGE2 using an assay kit (Amersham Pharmacia Biotech, NJ, U.K.). Briefly, chondrocytes were seeded in standard 96-well microtiter plates at 2×104 cells/well. Following addition of the indicated pharmacological reagents, supernatant was used to quantify the amount of PGE2, according to the manufacturer's protocol. PGE2 levels were calculated against a standard curve of PGE2 and normalized against the amount of genomic DNA.
Transfection and reporter assays
The reporter construct (3xPPRE-TK-luc/pGL3) used in the transfections contained three copies of the peroxisome proliferator response element (PPRE) site from the aP2 enhancer (ARE7) inserted upstream of a minimal thymidine kinase (TK) promoter in the pGL3 basic luciferase vector and RXRα cDNA (a gift from Dr. JB, Kim at Seoul National University). Transfection of the reporter vector was performed as described previously (23). Chondrocytes cells were grown in 10% fetal calf serum/DMEM and co-transfected with various reporter plasmids (1 µg) and pCMV β-galactosidase plasmid (0.5 µg) (Clontech, Palo Alto, CA, U.S.A.) using Lipofectamine (Life Technologies, Inc., Grand island, NY, U.S.A.), following the procedure recommended by the manufacturer. The transfected cells were cultured in complete medium for 24 hr and used for further assay. Cells were treated with increasing concentrations (0-20 µM) or times (0-24 hr) of 15d-PGJ2. Luciferase and β-galactosidase activities were determined in all extracts using a Luciferase assay (Promega, Madison, WI, U.S.A.) and Galacto-light system (Tropix, Inc., Bedford, MA, U.S.A.). The activities were corrected for transfection efficiency with β-gal activity and expressed as fold-induction relative to the level in untreated cells. Each assay was performed in triplicate. Data are the mean +/- SD of three separate experiments performed with cells from different animals (**pd 0.01 versus non treated cells).
Reverse transcriptase polymerase chain reaction (RT-PCR)
Primary culture chondrocytes were treated with 15d-PGJ2. Total RNA was isolated using RNA STAT-60 (Tel-Test B, Inc., Friendswood, TX, U.S.A.) and reverse-transcribed with MoMLV-RT (Invitrogen, California, U.S.A.) as previously described (26). The following primers and conditions were used for PCR in rabbit articular chondrocytes: for type II collagen (370-bp product), 5'-GAC CCC ATG CAG TAC ATG CG-3' (sense) and 5'-AGC CGC CAT TGA TGG TCT CC-3' (antisense) with an annealing temperature of 55℃; for SOX-9 (386-bp product), 5'-GCG CGT GCA GCA CAA GAA GGA CCA CCC GGA TTA CAA GTA C-3' (sense) and 5'-CGA AGG TCT CGA TGT TGG AGA TGA CGT CGC TGC TCA GCT C-3' (antisense) with an annealing temperature of 62℃; for COX-2 (298 bp product), 5'-TCA GCC ACG CAG CAA ATC CT-3' (sense) and 5'-GTG ATC TGG ATG TCA GCA CG-3' (antisense) with an annealing temperature of 52℃ for glyceraldehydes 3-phosphate dehydrogenase (GAPDH; 299-bp products), 5'-TCA CCA TCT TCC AGG AGC GA-3' (sense) and 5'-CAC AAT GCC GAA GTG GTC GT-3' (antisense) with an annealing temperature of 50℃ The primers for rabbit type II collagen and rabbit SOX-9 were de- signed based on the sequence of human homologs, and sequencing of PCR product showed 89% and 93% homology with corresponding human genes.
Data analyses and statistics
The results are expressed as the means±S.E. values calculated from the specified number of determinations. Analysis of variance (ANOVA) was performed to compare individual treatments with their respective control values. The Turkey's honestly significant difference method was employed when making multiple comparisons among means (24).
RESULTS
15d-PGJ2 induces dedifferentiation in articular chondrocytes
To examine the effects of 15d-PGJ2 on articular cartilage chondrocyte differentiation, rabbit articular chondrocytes in primary culture were treated with 10 µM 15d-PGJ2 for 48 hr. Under these experimental conditions 15d-PGJ2 displayed no cytotoxicity, as assessed by the conversion of a tetrazolium compound to its formazan product, as well as trypan blue exclusion (data not shown). 15d-PGJ2 inhibited type II collagen, a marker for differentiation of chondrocytes, and SOX-9 expression, a major transcription factor that regulates type II collagen expression, in a dose- and time-dependent manner as determined by immunoblot analysis (Fig. 1A). RT-PCR also showed that expression of type II collagen and SOX-9 was reduced (Fig. 1B). Similarly, 15d-PGJ2 treatment of primary culture cells blocked the accumulation of sulfated proteoglycan in a dose- and time-dependent manner (Fig. 1C, D). The effects of rosiglitazone, a PPAR-γ natural ligand, on dedifferentiation and COX-2 expression in articular chondrocytes showed the same results (data not shown). These results indicate that 15d-PGJ2 induces dedifferentiation of articular chondrocytes in primary culture cells.
15d-PGJ2 increases COX-2 expression and PGE2 production in articular chondrocytes
The effect of 15d-PGJ2 on COX-2 expression and PGE2 production was investigated using primary culture chondrocytes. In chondrocytes, 15d-PGJ2 increased protein levels of COX-2 in a time- and dose-dependent manner as determined by immunoblot analysis (Fig. 2A). COX-2 expression was increased 1 hr after 15d-PGJ2 treatment, and the levels peaked at 6 hr. 15d-PGJ2 treatment, however, did not affect PPAR-γ expression levels. 15d-PGJ2 also enhanced COX-2 expression as determined by RT-PCR (Fig. 2B). Consistent with the induction of COX-2 expression, 15d-PGJ2 stimulated PGE2 production in a time- and dose-dependent manner (Fig. 2C, D). The production of PGE2 was determined by using a PGE2 assay kit. PGE2 levels were calculated against a standard curve of PGE2. These data indicate that 15d-PGJ2 not only causes dedifferentiation of articular chondrocytes but also stimulates COX-2 expression and PGE2 production.
15d-PGJ2 causes dedifferentiation and induction of COX-2 expression in cartilage explants
To examine the effects of 15d-PGJ2 on articular cartilage chondrocyte dedifferentiation and COX-2 expression, cartilage explant cultures were treated with 10 µM 15d-PGJ2 for 24 hr, and cartilage-specific matrix molecules were determined. 15d-PGJ2 caused a dramatic loss of type II collagen (Fig. 3, middle panel) and sulfated proteoglycans (Fig. 3, left panel) as determined by immunohistochemical staining and Alcian blue staining, respectively. However, 15d-PGJ2 stimulated COX-2 expression (Fig. 3, right panel) as determined by immunohistochemical staining. These data indicate 15d-PGJ2 also induces dedifferentiation and COX-2 expression in cartilage explants.
15d-PGJ2 stimulates PPAR-γ-activation in articular chondrocytes
To determine whether the PPAR-γ expressed in the rabbit articular chondrocytes were transcriptionally active, a PPRE cloned upstream of luciferase (3xPPRE-TK-Luc/pGL3) was transiently transfected into articular chondrocyte cells, and the cells were then treated with 15d-PGJ2. As expected, expression of luciferase activity was significantly induced after 15d-PGJ2 treatment in a time- and dose-dependent manner (Fig. 4). The transfection efficiency was above 40% efficiency. These results indicate that 15d-PGJ2 increased PPAR-γ transcription activity in articular chondrocytes.
15d-PGJ2 regulates on dedifferentiation via PPAR-γ-dependent pathway but not COX-2 expression
Because 15d-PGJ2 in articular chondrocytes causes both dedifferentiation and COX-2 expression/PGE2 production, we next examined whether PPAR-γ activation is associated with dedifferentiation and/or COX-2 expression and PGE2 production. 15d-PGJ2-induced inhibition of type II collagen and SOX-9 expression was prevented by the addition of 10 µM BADGE (bisphenol A diglycidyl ether, PPAR-γ antagonist) (Fig. 5A). The 15d-PGJ2-induced inhibition of sulfated proteoglycan on primary culture chondrocytes was completely recovered by BADGE pretreatment (Fig. 5B). BADGE did not affect 15d-PGJ2-stimulated COX-2 expression (Fig. 5A) or PGE2 production (Fig. 5C). To elucidate the dedifferentiation mechanism, we examined PPAR-γ-response element (PPRE)-luciferase activity. As shown in Fig. 5D, 15d-PGJ2-enhanced PPAR-γ activity was blocked by BADGE pretreatment. On the basis of these results, we suggest that 15d-PGJ2 regulates dedifferentiation via a PPAR-γ-dependent pathway and COX-2 expression/PGE2 production via a PPAR-γ-independent pathway.
DISCUSSION
Chondrocytes in normal articular cartilage are a unique cell type in that their differentiated phenotype is reversible. Chondrocyte phenotype is regulated by a balance of anabolic and catabolic molecular reactions that are involved in maintaining homeostasis of cartilage tissue (23). Differentiated chondrocytes lose their phenotype and transform into fibroblast-like cells upon exposure to soluble factors IL-1β (25), and nitric oxide (26) or during serial subculture in vitro (23). Such a destruction of homeostasis is believed to be involved in the pathophysiology of arthritis (27).
It was shown previously that 15d-PGJ2 has anti-inflammatory effects on both human chondrocytes (10) and rheumatoid synovial fibroblasts (28). However, the present study demonstrated the pro-inflammatory effect of 15d-PGJ2 in rabbit articular chondrocytes, suggesting a catabolic role of 15d-PGJ2 in cartilage. Thus far, controversy still remains regarding the role of 15d-PGJ2 in inflammatory responses (29). Indeed, the 15d-PGJ2 pathway has been shown to have anti- and pro-inflammatory effect (29), depending on experimental conditions and/or cell types.
PPAR-γ is constitutively expressed in articular chondrocytes as shown by immunoblot in Fig. 2A. The presence of the expression of the PPAR-γ in chondrocytes may provide a new insight in the understanding of the mechanisms which lead to the loss of cartilage homeostasis. Under inflammatory and pathologic conditions, however, PPAR-γ plays an important role. Activation of this nuclear receptor by 15d-PGJ2 has been shown to be critical for the induction of rheumatoid arthritis and osteoarthritis. PPAR-γ thus deserves consideration for clinical testing in degenerative cartilage diseases (30).
In this study, we demonstrated that an increase in PPAR-γ activities is required for 15d-PGJ2-induced dedifferentiation of rabbit knee joint articular chondrocytes. However, 15d-PGJ2 treatment PPAR-γ activity is not required for inflammatory responses (i.e. COX-2 expression/PGE2 production). Thus, 15d-PGJ2-induced COX-2 expression/PGE2 production is mediated by a PPAR-γ-independent pathway. In 1995, 15d-PGJ2 was reported to be a high-affinity ligand for PPAR-γ, and diverse of its effects have been attributed to the activation of this nuclear receptor (11). However, there is increasing evidence that 15d-PGJ2 has diverse PPAR-γ-independent effects (31). Therefore, further studies are necessary to elucidate inflammatory responses-related different signaling mechanisms.
In summary, current knowledge indicates that PPAR-γ plays a key role in the regulation of dedifferentiation in articular chondrocytes.



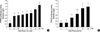
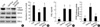
 XML Download
XML Download