

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Congenital pulmonary lymphangiectasis (CPL) is a poorly documented disease, and the disease is characterized by prominent lymphatic dilatation of septal, sub-pleural, and peribronchial tissue throughout both lungs. It is usually associated with congenital cardiac anomalies (1). Although there have often been debates on the classification and differential diagnosis of the disease, it can usually be divided into primary (congenital) and secondary forms (2-4). CPL is a uniformly fatal developmental defect that is related to an apparent failure of the communication between the pulmonary lymphatics and the systemic lymph channels. The primary form presents in neonates, and the patients mostly die due to the respiratory distress shortly after birth (1, 5). Based on the reported cases to date, most cases of CPL are diagnosed clinically or radiologically. However, there are rare cases that were pathologically confirmed on autopsy (1, 6-11). A review of the literatures published in Korean revealed three cases of CPL, and only one case was confirmed by lung biopsy (12-14). In this report, the authors performed an autopsy of a 13-day-old male neonate and a one-day-old male neonate, and confirmed pathologically that the sudden deaths of the neonates were due to CPL. Therefore, the authors report these cases, along with a review of the literature, since these are unique cases of CPL confirmed by autopsy.
MATERIALS AND METHODS
Case 1
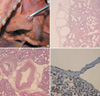
A male neonate with an unknown gestational and delivery history, who was the first baby born from non-consanguineous parents, suddenly died at the age of 13 days without any significant symptoms or signs while changing the diapers. The neonate weighed about 2,800 grams at birth. At autopsy, the baby measured 50 cm in length and weighed 2,852 grams. External examination revealed no specific abnormalities except several needle marks on the extremities and feces on the buttock area. On the internal examination, the left and right lungs weighed 42 grams and 50 grams, respectively. Both lungs were floated in the water. The pleural surface and inter-lobar space of both lungs showed numerous small, irregularly arranged nodules (Fig. 1A). After en bloc dissection of the thoracic organs, the lymphatic channels of paravertebral area were congested by lymphatic fluids. Under light microscopy, there was widespread ectasia of lymphatics in the pleura and in the interlobular septa, adjacent to pulmonary veins (Fig. 1B). These lymphatics were much larger than the neighboring veins, which was converse of the normal ratio. Dilated lymphatics were also found in the peri-bronchovascular bundle area (Fig. 1C). The remaining lung parenchyma was slightly thickened. The immunohistochemical stains for CD31 (1:50, DAKO, Denmark) and CD34 (1:50, DAKO, Denmark) were positive in the flat cells lining lymphatic space and blood vessels (Fig. 1D). D2-40 (1:100, DAKO, U.S.A.) was weakly and focally positive in the lymphatic endothelial cells. There were no more specific abnormalities in the lungs. The macroscopic examination of the heart revealed a small atrial septal defect, patent ductus arteriosus, and slight sub-adventitial hemorrhage of the pulmonary artery. On the histologic examination, the epicardial lymphatic spaces were slightly dilated. The kidneys and adrenals also showed mild lymphagiectasis. Other organs had no specific abnormalities.
Case 2
A one-day-old male neonate was admitted to the Department of Pediatrics due to cyanosis, moaning sound, and poor activity since birth. He was born by normal spontaneous vaginal delivery at 40 weeks of gestaion. His birth weight was 3,100 grams. The APGAR scores were 3 at 1 min and 4 at 5 min. At birth, crying was noted, but soon after cyanosis bation was performed, ventilator was connected, and surfactants were administrated. His skin color was returned to somewhat pinkish but changed to slightly cyanotic soon after. The initial chest radiography showed a reticulo-nodular pattern on both entire lung fields, and arterial blood gas analysis revealed severe hypoxemia. However, cyanosis and bradycardia were persistent, and pneumothorax of both lungs developed. Cardiopulmonary resuscitation was performed, but he expired on the same day. On maternal history, there was no specific history except premature rupture of membrane. On familial history, he was the first male baby. At autopsy, the baby measured 55 cm in length and weighed 3,480 grams. External examination revealed multiple needle marks on both extremities and two incision lines at the lateral chest for the closed thoracostomy. The skin was cold and moist. On the internal examination, the heart measured 4×3.5×2.2 cm, and the external contour was sweet potato-like appearance. The arrangement of the atrial chambers was situs solitus. Both pulmonary artery and aorta were arising from a single ventricle, and this ventricle looked like the right ventricle grossly. All pulmonary veins drained into the left atrium. The combined weight of both lungs was 110 grams. Both lungs were floated in the water. The pleural surfaces were carpeted with numerous tiny cystic spaces of variable sizes (Fig. 2A). Each lung had a normal lobation. On sections, there were numerous tiny cystic spaces within the lung parenchyma. On microscopic examination, there were widespread ectasia of lymphatics in the pleura, the interlobular septa, and peri-bronchovascular area (Fig. 2B, C). On immunohistochemical stains, the flat cells lining lymphatic space and vascular endothelial cells were positive for CD31 (1:50, DAKO, Denmark), and the lymphatic endothelial cells were weakly positive for D2-40 (1: 100, DAKO, U.S.A.) (Fig. 2D). There were also features of mild amniotic fluids aspiration, congestion, and focal intra-alveolar hemorrhage. The brain revealed diffuse subarachnoid hemorrhage and occipital intra-ventricular hemorrhage. Both kidneys revealed medullary interstitial hemorrhage. Other organs had no specific abnormalities.
DISCUSSION
Lymphatic channels are found throughout the human body and facilitate the transport of excess interstitial fluids from all tissues back into the cardiovascular system. Interference with the normal lymphatic passage results in excessive interstitial fluids accumulation, known as lymphedema. CPL is a part of a spectrum of lymphatic disorders (7). The significance of congenital abnormalities of the lymphatic system has been obscured by confusing terminology. In 1970, Noonan et al. (15) classified the pulmonary lymphangiectasis into three groups. Group 1 is a generalized form of lymphagiectasis (lymphedema with intestinal lymphangiectasis), group 2 is due to pulmonary venous hypertension or obstruction associated with cardiovascular anomalies, and group 3 includes patients compromised by a primary developmental defect of the pulmonary lymphatics. Lymphangiomas, lymphangiomatosis, lymphangiectasia, lymphedema, as well as these terms are frequently used interchangeably (2).
Recently, Faul et al. (4) proposed a new classification of these diseases, and the classification was based on the clinical presentation and pathologic features, rather than the assumed pathophysiology. They also divided the lymphangiectasis into the primary (congenital) and secondary forms to differentiate it from the lymphangiomatosis, and they noted that the primary form presents in neonates and is usually fatal. The secondary form of lymphangiectasis results from a variety of processes that impair lymphatic drainage and increase lymph production. They proposed that primary and secondary lymphangiectasis can be distinguished by the age of the patients and their clinical courses. Case 1 is a pulmonary lymphangiectasis without the presence of abnormalities in the cardiovascular system. This case is classified as the group 3 based on the Noonan's classification (15), and based on the recent classification by Faul et al. (4), this case can be classified as the primary pulmonary lymphangiectasis. Case 2 is a pulmonary lymphangiectasis with a single ventricle and normal pulmonary venous return. It can also be classified to primary pulmonary lymphangiectasis and group 3, based on the Noonan's classification (15).
CPL probably results from a failure of pulmonary interstitial connective tissues to regress, leading to the dilation of pulmonary lymphatic capillaries. It usually occurs after the 16th week of fetal life (4, 16). The secondary form can occur when surgery, radiation, infection, tumor, or trauma disturbs effective lymphatic drainage. Children with congenital heart disease, as well as adults with severe mitral valve disease, have an increased lymphatic circulation that contributes to the lymphatic dilatation and to the severity of lymphangiectasis (4, 15). CPL is commonly a sporadic occurrence, but a few cases of familial CPL have been reported (17). A case of CPL with 46,XY/46,XX mosaicism on chromosomal analysis was reported in Noonan Group 3 CPL. These XX/XY mosaicism cases may be associated with aggressive clinical manifestations and perinatal death (10, 18).
Macroscopically, the lungs of pulmonary lymphangiectasis appear heavy and noncompliant. The visceral pleura show numerous small dilated lymphatics, occasionally simple cystic space. The interlobular septa are widened and prominent. The lymphatic vessels were dilated or cystic in some instances (4). A small amount of collagen and smooth muscle may be found in the walls of vessels, particularly in the secondary form of pulmonary lymphangiectasis (3). Flat cells lining the lymphatic spaces are immunohistochemically stained for CD31, and this process indicates their endothelial nature, and it excludes the diagnosis of interstitial emphysema. In pulmonary emphysema, the spaces may be devoid of lining cells but may be connected in some areas with alveoli. The immunohistochemical staining for cytokeratin also showed the pneumocytes, lining intact alveolar walls. Interstitial emphysema can be distinguished from congenital cystic adenomatoid malformation because in interstitial emphysema there are areas of normal lung between the cysts. By contrast, adenomatoid malformation is composed of abnormally arranged immature lung, in which the cysts are usually lined with prominent epithelium including goblet cells (19). Our cases show mild thickening of interstitium of the alveolar walls by collagen, fibroblasts, and some inflammatory cells. Since such interstitial nonspecific changes can be seen in normal lungs or other case reports of CPL (1, 7, 8), it can easily be differentiated from other interstitial lung disease, such as interstitial pneumonia. In the case 1, the ante-mortem clinical data of this neonate could not be obtained. He died suddenly on the 13th day of birth with mild respiratory symptoms. Therefore, the definite diagnosis of CPL was obtained only by autopsy. Post-mortem examination of lungs revealed the sub-pleural and peri-bronchovascular small cystic spaces with flattened one-layered lining cells, and lymphatic dilatation with lack of considerable interstitial thickening. Mild lymphatic dilatation of heart, kidney, and adrenals was also present. Therefore, the interstitial pneumonia, interstitial emphysema, and congenital cystic adenomatoid malformation could be excluded.
It is also occasionally difficult to differentiate pulmonary lymphangiectasis from lymphangiomatosis by histological examination because both conditions have similar clinical manifestations and histological features. Tazelaar et al. (3) proposed that the term "lymphangiectasis" be reserved to describe those extremely unusual congenital or secondary lesions in which the primary alteration is a dilatation of existing lymphatic channels, without an increase in their number or complexity, while the term "lymphangiomatosis" be used for those diffuse lesions characterized primarily by an increased number of complex anastomosing lymphatic channels in which dilatation is a secondary phenomenon. Lymphangiomatosis and CPL share a similar immunohistochemical profile for vimentin, factor VIII-related protein, CD31, CD34, and smooth muscle actin. CD31 and CD34 display the most uniform pattern of endothelial reactivity. However, the use of these immunoprofiles cannot differentiate between lymphangiomatosis and CPL (7). Histological findings of the lungs in our cases reveals diffuse dilatation of existing lymphatic channels rather than an abnormal increase in their number or complexity. Therefore, the possibility of lymphangiomatosis can be ruled out. Recently, it was reported that another monoclonal antibody D2-40 is a highly sensitive and specific marker of lymphatic endothelium in normal tissue and vascular tumor (20). Our cases also show positive reactivity for CD31 and CD34 along the endothelial cells lining the cystic space and adjacent blood vessels. However, D2-40 was stained only in the lymphatic endothelial cells with weak intensity, showing a good contrast with CD31 and CD34.
Primary pulmonary lymphangiectasis is often fatal in early life, and the cases lead to frequent stillborn. Secondary pulmonary lymphangiectasis can present with respiratory distress at any age. Cases associated with pulmonary venous obstruction or other congenital heart defects, usually present during the early childhood. In addition, a number of congenital and genetic diseases have been associated with pulmonary lymphangiectasis, such as Noonan syndrome and Down syndrome (15). Primary pulmonary lymphangiectasis present soon after birth and is commonly fatal in early life. The dilated lymphatics, in association with chylothoraces, lead to life-threatening pulmonary hypoplasia and respiratory failure. Secondary lymphangiectasis presents as localized or diffuse pulmonary interstitial infiltrates or cystic lesions on chest radiography and magnetic resonance imaging (4). High-resolution computed tomography demonstrates extensive bilateral septal and peri-bronchovascular interstitial thickening, areas of ground-glass attenuation, and bilateral pleural effusions (11). However, a confirmative diagnosis can be made by histological examination (3).
Although several studies have been carried out on CPL, the pathophysiologic mechanism of CPL has not been clearly identified, except the physical mechanism, such as lymphatic obstruction. There is a need of further studies involving the molecular biological or genetic methods. Many patients with CPL die at their neonatal period or infantile period, as shown in these cases. The authors consider that such mode of death should also warrant further investigation. In general or forensic field of medicine, CPL is not a common disease. However, the progress of death due to this disease is very swift, and the identification of CPL is very difficult in the neonatal or infantile period. Therefore, it is necessary for forensic pathologists to be aware of the typical clinical manifestations, gross findings, and histological findings. The authors believe our cases will be of great help in performing the legal autopsy of similar cases.

 XML Download
XML Download