

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The syndromes of resistance to thyroid hormone (RTH) are characterized by reduced clinical and biochemical manifestations of thyroid hormone action relative to the circulating hormone levels (1). Since Refetoff et al. initially identified this syndrome in 1967 (2), over 1,000 cases have been identified (3). RTH has been associated with mutations in the thyroid hormone receptor-β (THRB) gene. These mutations have, been identified in RTH patients from 300 families, and 122 different mutations have been discovered (3). In 10% of cases of thyroid hormone hyposensitivity, no mutations can be detected in the THRB gene (4).
Only three cases (5, 6) of RTH have been reported in the Korean literature. All of these reported cases of RTH have been determined to be the result of point mutations in different regions of the THRB gene. One of these cases presented as pituitary RTH (5), and the other two presented as generalized RTH (5, 6). However, no insertion mutations have, been described in Korean RTH patients. Therefore, this report represents the first observation of generalized RTH associated with an insertion mutation in the THRB gene in Korea, found in two individuals from the same family. These results were confirmed via sequence analysis.
CASE REPORT
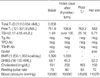
A Korean woman initially noticed an enlargement in her thyroid gland at the age of 43. The thyroid function test revealed elevated free thyroxine (T4) with nonsuppressed thyroid stimulating hormone (TSH). She exhibited none of the signs and symptoms of thyrotoxicosis, which include wasting, agitation, and tachycardia despite high serum levels of thyroid hormone. Ultrasonography revealed a diffuse goiter with a nodule in each lobe. Due to the findings of elevated serum thyroid hormone levels, coupled with increased thyroid radioiodide uptake (46% at 24 hr), the patient was presumed to have primary hyperthyroidism. Therefore, methimazole therapy was administered, and antithyroid drug treatment continued for several months. However, as the patient's TSH levels remained slightly elevated, and free T4 and triiodothyronine (T3) were still in excess of upper normal levels after antithyroid drug treatment, RTH was suspected, and so the methimazole treatment was discontinued. TSH-secreting adenoma was ruled out on the basis of the following findings: clinical phenotype without manifestations of thyrotoxicosis, a negative magnetic resonance imaging (MRI) of the pituitary gland, and the level of the -subunit of TSH being within normal range. Her serum level of free T4 was 751.6 pM/L (normal range, 121-322.8), total T3 was 0.058 nM/L (normal range, 0.012-0.034), and TSH was 1.49 mU/L (normal range, 0.17-4.05). Thyroid hormone and TSH levels were measured via radioimmunoassay (RIA) and immunoradiometric assay (IRMA). Thyroid autoantibodies, including anti-thyroid peroxidase antibody, anti-thyroglobulin antibody, and TSH receptor antibody, were all found to be negative. Basal metabolic rate, serum cholesterol, and serum sex hormone binding globulin levels were all normal. She exhibited resistance to thyroid hormone in peripheral and pituitary tissues. A polymerase chain reaction (PCR) sequencing of the THRB gene of the patient was subsequently conducted. Genomic DNA was isolated from the peripheral blood leukocytes of the patient, in accordance with the standard protocols. PCR amplification (PTC-200, MJ Research, Watertown, MS, U.S.A.) was conducted on seven coding exons (exon 4 to 10) of the THRB gene. The PCR products were then employed in a direct sequence analysis, using primers identical to those used for PCR (Table 2), with a BigDye Terminator system, version 3.0 (ABI Systems, Forester, CA, U.S.A.). The sequences were analyzed using the computational software, ABI PRISM Sequence Analysis, version 3.7 (Applied Biosystems, Forster City, CA, U.S.A.). In the sequence analysis, we discovered a mutation in exon 10 of the THRB gene, specifically, cytosine inserted at position 1358/1359 (1358_1359insC, Leu454PhefsX11) (Fig. 1).
During the period in which this patient was still being evaluated with regard to RTH, the patient also underwent a subtotal thyroidectomy at another hospital, in which the right lobe was completely removed and the left lobe was partially removed. This procedure was necessitated due to the discovery of solid nodules in the thyroid, which, according to the results of fine needle aspiration cytology, appeared to suggest a follicular neoplasm. The actual pathologic finding of the tumor was benign adenoma. When the patient revisited our hospital, she had shown to manifest postprocedural hypothyroidism and hypoparathyroidism. She was then placed on thyroxine, calcium, and vitamin D. The thyroxine dosage was repeatedly adjusted on the basis of her serum free T4 levels, pre-surgical TSH level, clinical manifestations of thyroid hormone excess or deficiency, and tissue indices of thyroid hormone action, including basal metabolic rate or sex hormone-binding globulin levels.
The patient had two children. Her 18-yr-old son and 12-yr-old daughter also visited our clinic in order to have their thyroid functions evaluated. Her son was found to have elevated thyroid hormone levels, but was clinically euthyroid. His serum free T4 level was 583 pM/L (normal range, 121-321.8), and his TSH level was 6.71 mU/L (normal range, 0.17-4.05). The 99mTechnetium thyroid scan revealed diffuse enlargement of the thyroid glands, increased uptake, and a warm nodule. Ultrasonography revealed a diffuse goiter with a solid nodule in the left lobe, which, upon fine needle aspiration cytology, was suggestive of benign adenoma. Sera from the mother and her son evidenced identical abnormalities, with regard to the insertion mutation in nucleotide 1,358/1,359 in exon 10 (Fig. 1B, C). Her daughter, however, exhibited no abnormalities, with regard to both her thyroid function tests and her THRB gene sequence. Detailed clinical information of the index case and of her son are shown in Table 1.
DISCUSSION
RTH is an inherited condition induced by defects that reduce the responsiveness of the target tissues to thyroid hormone (1). The incidence of RTH is probably approximately 1 case per 40,000 live births (7). The clinical presentation of RTH is also known to be highly variable. Whereas most individuals are clinically euthyroid, some individuals with RTH may appear to be overtly hypothyroid, while others may appear thyrotoxic. Furthermore, the same subject with RTH can manifest signs and symptoms of hypothyroidism in one tissue, while in another tissue, the findings may be suggestive of thyrotoxicosis. RTH patients that appear eumetabolic or hypothyroid are considered to be suffering from generalized RTH (GRTH), and those with a hypermetabolic clinical presentation are generally referred to as having pituitary RTH (3). Finally, the occurrence of isolated peripheral tissue resistance to thyroid hormone was reported in a single patient (8).
Thyroid hormone receptors (TRs) are ligand-dependent transcription factors, which mediate the biological activities of T3. TRs are encoded for by the THRA and THRB genes, which are located on chromosomes 3 and 17, respectively (3). The alternative splicing of the primary transcripts results in the formation of four T3-binding proteins (β1, β2, β3, and α1), as well as two proteins that do not bind to T3 (α2 and α3) (9). Although THRA1 and THRB1 are ubiquitously expressed, THRA1 is expressed primarily in the heart, bone, and brain, whereas THRB1 is more abundant in the liver, kidney, and thyroid. THRB2 expression is limited to the pituitary, hypothalamus, retina, and inner ear, and THRB3 expression has been detected principally in the heart and kidney (7, 11). 90% of RTH subjects, when studied at the level of the gene, have been found to harbor mutations in the THRB gene. No mutations have so far been detected in the THRA gene (3, 9). The THRB gene consists of 10 exons (12); the aforementioned mutations are located in the last 4 exons, which code for the hinge region and the ligand-binding domain of the receptor. The majority of these mutations result in a reduction in T3-binding and/or an impairment in transactivation. The majority of the previously described mutations are diverse missense mutations that induce single amino acid substitutions. However, insertion or deletion mutations of the THRB gene sequence are quite rare, and have been observed in only a very few cases (13-16).
To the best of our knowledge, this is the second report in the literature to describe a cytosine insertion mutation at this position in the THRB gene. Although this insertion mutation is definitely the first such case in the Korean literature, a similar case was previously reported by Takeda et al. (16). They described this mutation in the thyroid hormone receptor, which was identified as a cytosine insertion at position 1644, resulting in a frameshift mutation (Leu454PhefsX11) in a 74-yr old woman presenting with GRTH. A screening of this woman's family members resulted in the detection of similar biochemical abnormalities in two other relatives. These appeared to be mutations identical to those described in this paper.
Familial occurrence of RTH has been documented in approximately 75% of cases (3). Inheritance is autosomal dominant (3). Transmission was clearly recessive in only one family (2, 17). Consanguinity in a family with dominant inheritance of RTH has produced a homozygous child with severe resistance to the hormone who died at the age of 7 yr (18). Patients exhibiting a frameshift mutation from codon 454 to 463 can be considered to have GRTH, and appear to transmit the condition in an autosomal dominant manner, according to two reports (the present case, 16).
Another interesting characteristic of our two cases involves the thyroid nodules found prior to surgery. Thyroid enlargement in RTH patients tends to be diffuse, whereas nodular changes and gross asymmetry are more common features of recurrent goiters after surgery (1). Taniyama et al. (19) has reported only one case of GRTH that was accompanied by a toxic multinodular goiter and thyroid microcarcinoma. A molecular examination of this case revealed a R429Q mutation in the THRB gene. This is one of the mutations that is usually associated with a pituitary resistance phenotype. In our case, the solid nodule in the index case upon fine needle aspiration cytology was suggestive of follicular neoplasm. The postsurgical pathologic examination confirmed the existence of benign adenoma within the thyroid gland and found no evidence of malignancy. The etiology of nodular goiters has yet to be clearly elucidated, but continuous growth stimulation by thyrotrophic substances including TSH, thyroid growth stimulating immunoglobulin, and IGF-1, is believed to be involved in cell proliferation (20). Continuous stimulation by excess TSH may be a causative factor in some cases of RTH.
In this study, we have described a 1358-1359 insC mutation in exon 10 occurring in two members of the same family, both of whom were diagnosed with generalized RTH. This mutation has not been observed in any other RTH patients in Korea, but has been postulated to be the cause of RTH in the family described in this study.


 XML Download
XML Download