

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Limb girdle muscular dystrophies (LGMDs) are a clinically and genetically heterogeneous group of disorders, characterized by progressive weakness of proximal limb girdle muscles. To date, at least seven different forms of autosomal dominantly-inherited LGMD (LGMD1) have been recognized, and 11 in cases of autosomal recessively-inherited ones (LGMD2). LGMD2A (MIM# 253600) consists of 30-60% of all LGMD, and is assumed to be a major form among several ethnic groups (1-7). It is caused by single or small nucleotide changes distributed throughout a gene, named CAPN3 (MIM# 114240), which encodes the calpain 3 protein (8).
Calpains or calcium-activated neutral proteases are non-lysosomal intracellular cysteine proteases. The mammalian calpains include two ubiquitous proteins, calpain 1 (MIM# 114220) and calpain 2 (MIM# 114230), as well as two stomach-specific proteins, and calpain 3, which is muscle-specific. Unlike dysferlin and sarcoglycans, which are involved in LGMD2B and LGMD2C through 2F, respectively, calpain 3 is not a structural protein and disappears very rapidly in vitro due to autolysis (9), frustrating immunohistochemical staining popular in other muscular dystrophies. The human CAPN3 covers a genomic region of more than 40 kb and is composed of 24 exons. It is expressed predominantly in the skeletal muscle tissue as a 3.5 kb transcript, driving the translation of a 94 kD protein.
LGMD2A and the mutations in CAPN3 have not been studied in the Korean population, although it is expected to account for a large proportion of LGMD considering 26% in the Japanese population (6). Here we first report the results of mutational analysis of LGMD2A screened from Korean LGMD patients.
MATERIALS AND METHODS
Selection of the subjects
We selected 35 patients registered in our clinic from 2000 to 2004 with the clinical diagnosis of LGMD, who satisfied the following conditions: slowly progressive mild to moderate limb girdle weakness and elevated serum creatine kinase (CK) level, and various degrees of dystrophic changes on muscle biopsy. The age distribution was in the range of 12-56 yr old. Immunohistochemical stainings for dystrophin, merosin, dysferlin and sarcoglycans revealed one case of Duchenne muscular dystrophy (DMD; MIM#310200) in carrier state, one case of merosinopathy (MDC1A; MIM#607855), and seven cases of dysferlinopathy (LGMD2B; MIM# 253601), leaving 26 patients to be studied with western blot analysis.
Western blot analysis
In each patient, 10-20 pieces of 6 µm-thick muscle cryosections were obtained, homogenized in 60 µL of sodium dodecyl sulfate (SDS) sample buffer by sonication, and heated at 95℃ for 5 min. Two microliters from each sample was loaded to 6% SDS-polyacrylamide gel, and electrophoresed over 20 mA for 1 hr before transferred to polyvinylidene difluoride (PVDF) membrane. The blot was labeled with three kinds of primary antibodies each targeting different epitopes of calpain 3 (Calp3c/11B3, Calp3c/11A2, Calp3d/2C4; Novocastra, U.K.) (10) followed by secondary antibody reaction (Histofine Simple Stain Max Po, Japan) overnight and detection with ECL western blotting detection reagent (Amersham Bioscience, Piscataway, U.S.A.).
Western blotting for dysferlin was then performed on the patients with defective signal on at least one kind of calpain 3 antibody, to exclude secondary calpain 3 deficiency due to dysferlin deficiency (11).
Mutational analysis
In the patients with defective calapin 3 on western blot study, further analysis was performed by screening mutations in CAPN3. The purpose and methods were explained in detail and written informed consents were obtained prior to the study using their RNA and DNA. In order to identify mutations, RT-PCR of CAPN3 and direct sequence analysis were performed. Six sets of primers described before (12) were used to cover the entire coding sequence of CAPN3, split into about 600 bp-segments. Each PCR product was directly sequenced using BigDye terminator cycle sequencing kit (Applied Biosystems, Foster City, U.S.A.) and 3730xl DNA analyzer (Applied Biosystems). The aberrant sequences detected by sequence analysis were first searched in the Leiden Open Variation Database (13). For the two mutations not found in the database, PCR-RFLP assay or direct sequencing was performed on each 100 alleles from a normal Korean population to exclude normal variation. For c.2125T>C (p.709Ser>Pro), genomic DNA region was amplified with primers CAPN3-ex20.a (5'-GGGGATTTTGCTGTGTGCTGT-3') and CAPN3-ex20.m (5'-ATTCCTGCTCCCACCGTCTC-3') (8). The products were digested by restriction endonuclease Hha III (Takara Bio, Tokyo, Japan) for 2 hr in 37℃, to make a pattern of 234 bp and 106 bp in wild type, in contrast to the bands of 234 bp and 53 bp in mutant. For c.2355-2357delTTC (p.786delPhe), we could not find an appropriate restriction endonuclease that we amplified exon 22 by PCR using primers CAPN3-ex22.a (5'-CACAGAGTGGCCGAGAGGCA-3') and CAPN3-ex22.m (5'-GGAGATTATCAGGTGAGATGCC-3') (8), and performed direct sequencing.
Clinical analysis of patients with calpain 3 deficiency
The clinical features of the patients, who showed calpain 3 deficiency on western blot analysis, were reviewed retrospectively based on their clinical records. The clinical history was obtained from each patient in detail with the special interest on the age at onset, first clinical symptom, the speed of progression, current disabling problems, and family history. For clinical examination, presence of muscle atrophy or hypertrophy, and associated musculoskeletal deformities were carefully inspected. We examined changes in their tendon reflexes and individual muscle power according to the Medical Research Council (MRC) grade at first presentation, and at certain points during clinical follow-up. The laboratory tests including complete blood count, liver and renal function tests, thyroid function test, chest radiography, electrocardiogram (ECG), serum electrolytes, and serum creatine kinase (CK) level were done in each patient. Muscle biopsies were obtained from biceps muscles by open biopsy, and were processed for routine histochemistry and reviewed for the diagnosis of muscular dystrophy.
RESULTS
Western blot analysis
Four patients were found to show defective signal with at least one primary antibody. Antibody Calp3c/11B3 was the most sensitive showing defective calpain 3 band in all four patients, while antibody Calp3c/12A2 showed defective bands in patient 1 and 4, and antibody Calp3c/2C4 only in patient 4 (Fig. 1). No patient with deficient calpain 3 was associated with dysferlin deficiency, supporting the evidence that all of them are primarily deficient for calpain 3. None of the others were deficient for either calpain 3 or dysferlin.
Mutation analysis
By direct sequencing of the CAPN3 mRNA amplified by RT-PCR, five different mutations were identified among the eight alleles, which are summarized in Table 1. We could find only one mutation in cDNA from patient 1 and none from patient 3. Patient 2 showed homozygous mutations. Of the pathogenic mutations identified, the missense mutation c.2125T>C (p.709Ser>Pro) from patient 1 (Fig. 2A) and the in-frame deletion c.2355-2357delTTC (p.786delPhe) from patient 4 (Fig. 2B) were novel, and no same change was observed among 100 control alleles using PCR-RFLP and the direct sequence analysis technique. Two other mutations (c.440G>C, c.1076C>T) had been reported elsewhere (6, 14).
Clinical features of the patients with calpain 3 deficiency
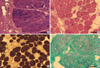
Patient 1, in whom we found a missense mutation (c.2125T>C [p.709Ser>Pro]) in one allele, was 19 yr old at the time of her first presentation to us. She developed difficulty in climbing upstairs and leg pain at age 12, which slowly progressed to running difficulty at age 15. At presentation, she could not lift up heavy objects or go upstairs without using handrails. Although she did not show waddling gait, a clear self-climbing motion was observed when she was asked to stand up from squatting posture. Neither calf pseudohypertrophy nor muscle atrophy was evident. On muscle power examination, she had MRC grade 3 weakness in leg elevation, and grade 4 in arm abduction, leg extension, leg adduction, leg abduction, knee extension, and knee flexion. The deep tendon reflexes were hypoactive symmetrically in limbs. She had no family history of similarly affected persons. Her serum CK level was elevated to 1,275 IU/L (normal; 5-275 IU/L), and other laboratory tests including ECG and chest radiography were unremarkable. Muscle biopsy showed marked muscle fiber size variation, increased connective tissue, clusters of regenerating fibers, and scattered whirled fibers with fiber splitting (Fig. 3A).
Patient 2, who had a homozygous missense mutation (c.1076C>T [p.359Pro>Leu]) at exon 8, was a 47-yr-old female at the time of her muscle biopsy. At her age of 30, she first noticed difficulty in climbing stairs, which progressed to limitations in daily activities by age 44. At presentation, she had difficulty in rising up from chair and could not stand up by herself from sitting position. Her muscle power was reduced to MRC grade 1 in leg adduction, grade 2 in hip flexion, hip extension, and leg abduction, grade 3 in shoulder elevation, elbow flexion, knee flexion and knee extension, and grade 4 in elbow extension and wrist flexion. Her deep tendon reflexes were lost in limbs except for the ankle jerks. She had no affected family members, and her serum CK level was mildly elevated (496 IU/L). The ECG and chest radiography were normal. Muscle biopsy showed a marked fiber size variation, markedly increased adipose connective tissue, many fibers with internal nuclei, scattered necrotic and regenerating fibers, some fibers undergoing fiber splitting, and type I fiber predominance (Fig. 3B, C).
The patient 3 had been wheelchair-bound for one year when he first presented to us at age 35. He became symptomatic with gait and running difficulty when he was 13 yr old. Since then, his disease was progressively worsened. He started to use canes at age 32 and became wheelchair-bound at age 34. He had a profound muscle weakness on examination. His muscle power was graded as 1 in leg elevation, leg extension, leg adduction, and leg abduction, 2 in distal leg muscles, 3 in upper extremity muscles, and 4 in quadriceps muscles. He also had generalized muscle atrophy, especially in shoulder, pelvic girdle, and intrinsic hand muscles. His deep tendon reflexes were lost except for the knees. He had no affected family members, and serum CK was slightly elevated to 500 IU/L. His ECG and chest radiography were unremarkable. Although he showed loss of immunoreactivity against all the three sets of anti-calpain 3 antibodies, we were not able to find any CAPN3 mutations. His muscle biopsy showed a marked fiber size variation, markedly increased adipose connective tissue, many fibers with internal nuclei, scattered necrotic and regenerating fibers, many fibers with rimmed vacuoles, and type I fiber predominance (Fig. 3D).
Patient 4, who was compound heterozygous for the mutations c.440G>C (p.147Arg>Pro) and c.2355-2357delTTC (p.786delPhe), was at her age of 21 when she first visited us with progressive difficulty in climbing stairs and rising from chairs. She became symptomatic when she was 8 yr old with running difficulty and tiptoe gait. The tenorrhaphy had been repeatedly performed due to progressive Achilles contracture. At present, she has gait disturbance with waddling, difficulty in climbing stairs and rising from chairs. Her muscle power was significantly reduced to MRC grade 1 in leg adduction and grade 2 in leg elevation. Muscles in proximal upper limbs and all other lower limb muscles were less severely affected and were graded as 4 to -4. She had a generalized areflexia. Her familial history was significant for her brother and sister who had the same problems with her. Her serum CK level was elevated to 1,104 IU, and ECG and chest radiography were normal. In muscle biopsy, there was a moderate fiber size variation, mildly increased adipose connective tissue, and a few regenerating fibers.
DISCUSSION
This is the first attempt to analyze the CAPN3 mutations among Korean LGMD patients. We identified four patients with calpain 3 deficiency by western blot analysis, and finally found four different mutations including two novel ones. One of our new mutation, c.2125T>C, changes a codon for polar amino acid serine into nonpolar proline, which is likely to produce a conformational change in the protein structure of calpain 3. Another new mutation c.2355-2357delTTC produces a deletion of single amino acid phenylalanine at codon 786. In multiple alignment analysis, both mutations are located in highly conserved regions of amino acid sequences across several species (Fig. 4), strongly supporting that they are pathogenic mutations. It is also remarkable that c.440G>C mutation had been previously reported in a Japanese population, which is known to be in close ethnic proximity with the Korean population.
In contrast to other reports (1-7), we could find just a small number of patients with CAPN3 mutations from a LGMD patient pool. This is thought to be partly because we could not reliably exclude autosomal dominant cases, but also that we have screened the patients first by western blot analysis. Although western blot analysis is still considered as a standard tool for the screening of LGMD2A, it seems neither perfectly sensitive nor specific (10, 15-17). According to a study employing a large number of genetically confirmed LGMD2A patients, the diagnostic sensitivity and specificity of western blot analysis were estimated as 52.5% and 87.8%, respectively (17). In our study, we used three types of western blot antibodies in order to maximize the sensitivity, but the antibody Calp3c/11B3 was found to be the most useful, which is known to have the widest reaction profile, binding other calpain molecules even from other species (10). Direct sequencing of the CAPN3 gene cannot detect all the mutations either, as exemplified by our case, that the two techniques are complementary. It is contradictory that patient 3 in whom we could not find any pathologic mutation, showed the clearest pattern of calpain 3 abnormality on western blot. Overall, we have failed to detect mutations in three alleles. As we have sequenced CAPN3 by RT-PCR, each segment spanning about 600 bp, a large deletion involving primer site could be the cause, although such mutation has not been reported so far. Mutations may be in the regulatory region, affecting the transcriptional activity, which has not been reported either.
The clinical features of LGMD2A had been studied and characterized in a few previous studies, and following features are now considered as typical: onset between 8-16 yr, early symmetrical weakness of proximal legs, no facial or cardiac involvement, normal intelligence, wheelchair-bound between 20 to 40 yr old, no muscle contracture except for the Achilles tendon, hyporeflexia, and a 5-20 fold increase in the CK level (16, 17, 19, 20). The clinical features of our patients are considered to be typical for LGMD2A. All of our patients showed a high CK level ranging 496-1,275 U/L. The weakness was predominant in their lower extremities, the onset was from childhood except for patient 2, deep tendon reflexes were lost or reduced, and none had mental or cardiac abnormalities.
Although LGMD2A is supposed to account for a major proportion in LGMD, it has been neglected in the Korean populations, partly due to the costly diagnostic procedure. Further studies using a larger number of patients with LGMD2A are needed to fully elucidate the genotypic and clinical characteristics of Korean patients with LGMD2A.




 XML Download
XML Download