

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cataplexy is a symptom that occurs when the muscle tension in various areas of the body is suddenly decreased involuntarily and lasts from few seconds to several minutes. It is a pathognomonic symptom of narcoplepsy, generally induced in response to laugh, excitement, anger, and other emotional changes (1, 2). In addition, among excessive daytime sleepiness, cataplexy, hypnagogic hallucination, sleep paralysis and nocturnal sleep disorder, which are known to be a symptom pentad of narcolepsy, cataplexy has been known to be a prerequisite clinical symptom required for the diagnosis of narcolepsy according to the International Classification of Sleep Disorders (3). In the absence of cataplexy, the diagnosis of broad-spectrum narcolepsy is possible by the presence of other clinical symptoms of narcolepsy and the result of multiple sleep latency test. On the other hand, for the diagnosis of narrow-spectrum narcolepsy, the presence of cataplexy is essential and the narcolepsy with cataplexy is considered to be an etiologically homogeneous disease. Especially, in cases where cataplexy is either clinically typical or severe, the frequency of HLA-DQB1*0602 has been shown to be higher (4, 5).
Hypocretin is a pair of neuropeptides discovered in the hypothalamus in 1998, consisting of hypocretin-1 (Hcrt 1) and hypocretin-2 (Hcrt 2), and it has been shown that they bind to the G protein-coupled receptors, hypocretin-1 receptor (Hcrtr 1), and hypocretin receptor-2 (Hcrtr 2), increasing intracellular calcium (6, 7). At the time of its discovery, hypocretin has been introduced as a neuropeptide that increases appetite and is involved in the regulation of body temperature, endocrine function, the cardiovascular system, etc. (8). More recently, the report that showed canine narcolepsy is caused by a mutation in the hypocretin-2 receptor gene (9, 10), and the report that showed behavioral patterns and the electroencephalographic findings in preprohypocretin knock-out white rats are similar to those of narcolepsy patients (11) draw attention to the association of hypocretin and narcolepsy. Subsequently, it has been shown that hypocretin nerve cells mediate excitatory action on monoaminergic neurons and thereby they are involved in the normal sleep awake cycle (12). In addition, it was reported that the expression of c-fos was increased in hypocretin neurons, during the awake period and the expression of c-fos is decreased in hypocretin neurons during the NREM sleep period, indicating the association of hypocretin and sleep regulation (13).
Until now, the mutation in the preprohypocretin gene or the hypocretin receptor gene in human narcolepsy has not been detected (14). However, it has been reported that in most narcolepsy patients, 80-90% of hypocretin cells in the hypothalamus were destroyed (15), and the transcription of preprohypocretin mRNA was significantly decreased in the brain of narcolepsy patients (14). Along with these findings, the report showing the concentration of hypocretin in the cerebrospinal fluid (CSF) was measured below the detection limit in over 80% narcoleptics (16, 17) suggests that hypocretin deficiency may be one of the most important pathophysiologic mechanisms of narcolepsy.
In this regard, this study was performed on narcoleptics diagnosed by polysomnography and multiple sleep latency test (MSLT) to understand the pathophysiology of narcolepsy in Korean narcolepsy patients by examining the frequency of HLA-DQB1 allele and the concentration of CSF hypocretin. In addition, this study was designed to investigate the frequency of the HLA-DQB1 allele and CSF hypocretin levels in Korean narcoleptics with cataplexy as compared with those who have not cataplexy.
MATERIALS AND METHODS
Subjects
From August 2003 to July 2005, among the patients reporting excessive daytime sleepiness, those who were suspicious of having narcolepsy were selected, based on diagnostic criteria for The International Classification of Sleep Disorders (3), and polysomnography and MSLT were performed at Sleep Disorders Clinic of St. Vincent's Hospital, The Catholic University of Korea. All patients with a chief complaint of excessive daytime sleepiness not obviously from sleep disordered breathing (SDB), delayed sleep-phase syndrome or behaviorally-induced insufficient sleep syndrome were included. Seventy-five subjects were selected, who had at least 2 sleep onset REM periods (SOREMPs) on MSLT, and the mean sleep latency shorter than 5 min. And in polysomnography, they did not have any evidence of sleep disorders that can cause excessive daytime sleepiness such as sleep apnea syndrome, periodic limb movement disorder, etc. Seventy-two of the seventy-five subjects (96%) accepted further evaluation, including blood and CSF tests.
Seventy-two narcoleptic patients underwent HLA typing to derermine the DQB1 allele and spinal tapping to derermine the level of CSF hypocretin. The study subjects were divided into a narcolepsy with cataplexy group (n=56) and a narcolepsy without cataplexy group (n=16) according to the presence or absence of cataplexy. Narcoleptics were excluded if they had a personal history of medical illness that may affect sleep, substance or alcohol abuse, seizure disorder, definite neurological deficit, and mental disorder that may cause sleep abnormalities. This study was approved by the institutional review board of the St. Vincent's Hospital, The Catholic University of Korea. Informed consent was obrained according to the Declaration of Helsinki.
Methods
All clinical symptoms of narcolepsy were collected through a structured interview in each patient by psychiatrists who had completed the sleep medicine course and through a sleep questionnaire. The structured interview revealed the frequency of excessive daytime sleepiness, cataplexy, sleep paralysis, hypnagogic hallucination, and the characteristic of cataplexy. In the questionnaire, triggering factors, duration, frequency of cataplexy were included, and the Stanford Center for Narcolepsy Sleep Inventory (18) consisting of total 146 questions was used.
Lymphocytes were isolated from the blood (2×106 cells/mL), and 0.5 mL PCR-K buffer (10× PCR buffer 1 mL, NP-40 40 µL, Tween-20 45 µL, proteinase K (20 mg/mL) 30 µL, D/W 8.8 mL) was added, and dissolved by treating at 58℃ for 60 min, and treated at 95℃ for 10 min, to inactivate proteinase K, and DNA was extracted. To determine the HLA-DQB1 allele, allele-specific probes were labeled with Dig-11-dUTP using terminal transferase, each sample was dropped on a Nylon membrane, Dig-11-dUTP labeled allele-specific probe was hybridized with the membrane dropped a sample, the expression was assessed using anti-DIG antibody, and finally the genotypes were determined.
Between 11 a.m. and 4 p.m., CSF was collected by lumbar puncture, and immediately stored frozen at -70℃ until the measurement of hypocretin. Hypocretin-1 was measured by 125I radioimmunoassay (RIA) kit (hypocretin-1: Phoenix Pharmaceuticals, Mountain View, CA, U.S.A.). CSF was acidified with the same volume of 0.1% trifluoroacetic acid (TFA), purified using equilibriated C-18 Sep-Columns (Phoenix Pharmaceuticals), and the columns were washed with 0.1% TFA twice and eluted with 3 mL of the mixture of 0.1% TFA and 60% acetonitrile (HPLC grade). The eluted solution was dried by applying negative pressure, and the pellets produced were dissolved with 300 µL RIA buffer. The detection limit was 40 pg/mL, and all samples were measured in duplicate.
Statistical analyses
Statistical analysis was performed using SPSS for Windows (Version 10.0). Demographic data, the frequency of clinical symptoms, and the HLA-DQB1 allele frequencies were presented as the mean±standard deviation, as well as in percentage. These variables and the CSF hypocretin levels were analyzed by independent t-test and chi-square test. A p-value less than 5% was considered statistically significant.
RESULTS
Demographical characteristics
Of the total 72 patients, 47 were male patients (65.3%), and 25 were female patients (34.7%). The age distribution was 7-68 yrs (mean age 26.4±11.6 yrs). The mean body mass index was 24.2±3.7 kg/m2, and the mean duration of illness was 9.5±8.3 yr.
The narcolepsy with cataplexy group consisted of 34 male patients (60.7%) and 22 female patients (39.3%). The age distribution was 7 to 59 yrs (mean age: 25.4±10.4 yrs). The mean body mass index was 24.5±3.7 kg/m2, and the mean duration of illness was 9.9±8.8 yr.
The narcolepsy without cataplexy group consisted of 13 male patients (81.3%) and 3 female patients (18.8%). The age distribution was 8-68 yrs (mean age 29.8±14.8 yrs). The mean body mass index was 23.1±3.8 kg/m2. The mean duration of illness was 8.3±6.2 yr. No significant differences were observed in demographic variables between two groups (Table 1).
Clinical symptoms
Excessive daytime sleepiness was shown in all 72 patients (100%), and it was found that cataplexy was experienced in 56 patients (77.8%), hypnagogic hallucination in 40 patients (55.6%), and sleep paralysis was experienced in 40 patients (55.6%).
Hypnogogic hallucination was occurring more frequently in the narcolepsy with cataplexy group (36/56, 64.3%) than in the narcolepsy without cataplexy group (6/16, 25.0%) with a statistical significance (p=0.005). In addition, sleep paralysis was found to be experienced more frequently in the narcolepsy with cataplexy group, but the difference was not significant (34 patients among 56 patients [60.7%] vs. 6 patients among 16 patients [37.5%]) (Table 2).
CSF hypocretin level
The CSF hypocretin concentration was lower than 110 pg/mL in 54 of the total 72 patients (75.0%). The hypocretin levels were decreased (≤110 pg/mL) (19) or below the detection limit of assay (<40 pg/mL) in 48 out of 56 cataplexy-positive patients (85.7%). In comparison, only 6 of 16 cataplexy-negative patients (37.5%) had a decreased hyopcretin level (p<0.001) (Table 3).
HLA-DQB1 Allele
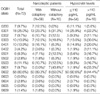
Of 72 narcoleptic patients, 58 patients (80.6%) had HLA-DQB1*0602. HLA-DQB1*0602 was significantly more frequent in cataplexy-positive patients (50/56, 89.3%) than in cataplexy-negative patients (8/16, 50.0%) (p<0.001). On the other hand, the frequency of HLA-DQB1*0601 was found to be significantly lower in the narcoleptics with cataplexy group (0 subjects, 0% vs. 7 subjects, 43.8%) (p<0.001) (Table 4).
The patients with a decreased CSF hypocretin level had a higher rate of HLA-DQB1*0602 (50/54, 92.6%) and a lower rate of HLA-DQB1*0601 (1/54, 1.9%) than those with a normal hypocretin level (8/18, 44.4% and 6/18, 33.3%, respectively) with a statistical significance (p<0.001 and p=0.001 for each comparison) (Table 4).
DISCUSSION
In this study, 77.8% of the narcolepsy patients experienced cataplexy, which is in line with previous studies reporting over 70% narcolepsy patients showed cataplexy (20, 21). As the main pathophysiology of cataplexy, the imbalance between monoaminergic and cholinergic transmissions in the pons has been proposed. In addition, the cholinoceptive site in the basal forebrain has been reported to be involved in a strong emotional stimuli (22, 23). During cataplectic episodes, muscle atonia is caused by suppression of motor neurons at the spinal level, together with the loss of deep tendon reflex, H-reflex, and autonomic nervous symptoms such as increased blood pressure and decreased heart rate (23, 24). During the initial period of cataplexy, the normal wake EEG is detected, however, when cataplexy prolongs, the EEG becomes more of REM sleep pattern, and the patient recalls a dream (25). Such observations support the proposal that cataplexy is an abnormal REM sleep phenomenon. In this study, narcoleptics with cataplexy experienced more hypnagogic hallucination, another REM sleep-related symptom, as compared with narcoleptics without cataplexy. This result is thought to be a finding that confirms the association of cataplexy and REM sleep, suggesting that the narcolepsy with cataplexy and without cataplexy may be heterogeneous groups.
As has been previously reported in other populations (4, 5, 26, 27), we also found that HLA-DQB1*0602, which has been reported to be positive in approximately 25% of the general population, was expressed 80.6% of the narcolepsy patient group. This is a result supporting the proposal that the HLA-DQB1*0602 positivity is a genetic marker for narcolepsy regardless of the racial backgrounds. In addition, the positivity of HLA-DQB1*0602 in the narcolepsy with cataplexy and without cataplexy was 89.3% and 50.0%, respectively. This result is also in agreement with previous reports that the HLA-DQB1*0602 positivity is higher in narcolepsy showing typical or severe cataplexy (4, 5, 27). In addition, we found that the HLA-DQB1*0601 allele was not expressed in the narcolepsy with cataplexy group, which is in agreement with the previous hypothesis that DQB1*0601 may serve as a protective gene against narcolepsy (28, 29). This HLA-DQB1 allelic differences between narcolepsy with cataplexy and without cataplexy may serve as the basis of the possibility that two groups may be etiologically heterogeneous.
Our study also investigated the CSF hypocretin levels in all participants. We found that the deficiency of CSF hypocretin was noted in 75% of Korean narcoleptic, which is a result similar to the previous studies (16, 17). In addition, similar to previous reports that showed a low concentration of hypocretin in 9 patients among 10 patients (90%) (17), 9 patients among 11 patients (81.8%) (30), and 23 patients among 26 (88.5%) (31), our study revealed 48 patients among 56 narcoleptics with cataplexy (85.7%) had a hypocretin concentration either low (≤110 pg/mL) or lower than the detection limit (40 pg/mL), while the concentration decreased only in 6 patients among 16 narcoleptics without cataplexy (37.5%). These results raise the possibility that hypocretin deficiency may be an important pathophysiologic mechanism in the development of narcolepsy and also support the fact that narcolepsy with cataplexy and narcolepsy without cataplexy may be different diseases regarding their causality. Also, in 54 narcolepsy patients with hypocretin deficiency, as compared with narcoleptics with a normal hypocretin level (n=18), the frequency of HLA-DQB1*0602 was found to be significant high (50 subjects, 92.6% vs. 8 subjects, 44.4%) and HLA-DQB1*0601 was found to be low (1 subjects, 1.9% vs. 6 subjects 33.3%). This suggests that the CSF hypocretin concentration might be related with HLA-DQB1 allele, and HLA-DQB1*0602 and HLA-DQB1*0601 might be have an important role in developing narcolepsy.
The limitations of our study are the absence of results from a normal control group was the small number of narcoleptics without cataplexy.
In summary, this is the first study in Korea that investigated the association of cataplexy with HLA-DQB1 allele and hypocretin level. The high HLA-DQB1*0602 positivity, DQB1*0601 negativity, and hypocretin deficiency in narcolepsy with cataplexy in our study confirms that narcolepsy with cataplexy and narcolepsy without cataplexy are genetically and pathophysiologically different disease entities.


 XML Download
XML Download