

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
A common cause of brain injury during the perinatal period is hypoxic-ischemic injury, which frequently results in the chronic handicapping conditions such as cerebral palsy, mental retardation, learning disability, and epilepsy (1). Predicting the outcome of neonates with hypoxic-ischemic injury, however, is difficult, because some methods that predict outcome have not reliably or consistently predicted long term neurologic outcomes. Recently, 1H-magnetic resonance spectroscopy (MRS) has been used as a quantitative noninvasive tool to assess the biomechanical changes associated with central nervous system injury. 1H-MRS, which provides objective extent of hypoxic-ischemic insults, enhances predictability and provides a method to assess treatment effect (2-5).
The exact mechanism by which hypoxic-ischemic brain injury occurs in neonates is not clear, although increasing evidences indicate that hypoxic-ischemic induced neuronal death includes both necrosis and apoptosis. Necrosis may predominate in acute damage, whereas apoptotic injury may take time to develop. Therefore, blocking the apoptotic cascade may prolong the therapeutic window after hypoxic-ischemic injury (6-8). There is evidence for involvement of multiple caspases in hypoxic-ischemic brain injury. And caspase inhibitors which were developed as antiapoptotic agents, are believed to play a key role in the delayed neuronal cell death observed after hypoxic-ischemic injury (7, 9).
The effects of growth hormone (GH) on the central nervous system have become more apparent in the past decade. Not only it is involved in brain growth and development, but its qualities as a neuroprotective factor against injury are now appreciated. Recent studies have demonstrated that GH is involved in neuroprotection during hypoxic-ischemic brain injury (10, 11). These protective roles are supported by the ability of GH to accelerate glial cell division and myelinogenesis, and GH is also thought to have neuroprotective roles in neurogenesis (11). Although this neuroprotective mechanism is not completely known, is probably achieved by inhibiting of caspase activities (12, 13).
The lipid peak in the 1H-MR spectrum has been reported to be a marker for apoptosis during hypoxic-ischemic injury (14). We have therefore used 1H-MRS to evaluate the effects of GH as a caspase inhibitor on hypoxic-ischemic injury in neonatal rat brains.
MATERIALS AND METHODS
Animals
The right common carotid arteries of 7-day old Sprague-Dawley rats (mean weight=13.3 g) were ligated under halothane anesthesia. After a recovery period of 3 hr, they were exposed to 8% oxygen at 37℃ for about 120 min. GH (Eutrophin, LGPhD, Korea) was administered just prior to hypoxic-ischemic insult. The rats were divided into four groups: control (10 µL distilled water, n=29), intracerebroventricular (ICV, 10 µL GH in 10 µL distilled water, n=23), intracerebroventricular/intraperitoneal (ICV+IP, n=21), and intraperitoneal (IP, 10 mg/kg GH in distilled water, n=23).
1H-MRS, TUNEL staining, and gross morphologic changes
Localized in vivo 1H-MRS was performed on a Bruker Biospec 4.7T MRI/MRS System equipped with active shielded gradients and ASPECT3000 computer with TOMIKON hardware and software (Bruker, Fallanden, Switzerland). Spectra were acquired in the right cerëbral hemisphere of rats 24 hr after the onset of hypoxic-ischemic insult. Water suppressed 1H-MR spectra were acquired using a VOSY sequence with detection of the double-refocused spin echo signal from the selected voxel (3×2×2 µL, 12 µL) using the following acquisition parameters: SW=5,000 Hz, SI=4,096 pts, NS=128, TR/TE=3,000/30 and 135 msec. To identify the peak at 1.3 ppm, the spectra were acquired at echo times of 30 and 135 msec in order to differentiate the lactate peak from the lipid peak. Peak areas were measured and the lipid/N-acetyl aspartate (NAA) and lipid/creatine (Cr) ratios were used as apoptotic markers.
After the 1H-MRS examinations on the 1st day, 6 brains from each group were perfused with 0.9% saline solution mixed with 2 units/mL of heparin, followed by perfusion with 4% paraformaldehyde in PBS solution. Each brain was isolated, and TUNEL staining was performed using an in situ Cell Death Detection Kit, POD (Boehringer Mannheim, Germany), as described. Apoptotic cells were counted 3 times in the parietal area of the brain using a ×200 lens, and the mean apoptotic cell numbers were calculated using Image Analyzer software.
Gross morphologic changes were scored at 2 weeks using a 5 point grading system as method by Palmer et al. (15), where 0 indicates no change and 4 indicates the most severe injury.
Statistical analysis
Data were expressed as mean±SD. Significance was assessed by unpaired t-test and ANOVA followed by Kruskall Wallis test. Spearman correlation was used to investigate relationships between lipid/NAA and lipid/Cr ratio and morphologic score. A p-value of 0.05 indicated statistical significance.
RESULTS
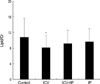
The lipid/NAA ratio was significantly lower in the ICV (8.5±3.1) and ICV/IP (9.2±2.5) groups than in the control group (12.2±4.6); although the lipid/NAA ratio in the IP group (11.4±3.7) was lower than in the control group, this difference was not significant (Fig. 1). The lipid/Cr ratio was also significantly lower in the ICV group (8.1±3.5) than in the control group (10.9±4.4); the lipid/Cr ratio was lower in the ICV/IP (9.2±3.4) and IP (9.6±3.5) groups than in the control group, but these differences were not statistically significant (Fig. 2).
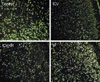
TUNEL staining was done on the 1st day. Although the number of TUNEL positive cells did not differ between the control and IP groups, there were fewer in the ICV and ICV/IP groups (Fig. 3).
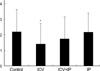
We scored gross morphologic changes at 2 weeks to evaluate the effects of GH. We found that the morphologic scores were significantly lower in the ICV group (1.4±1.3) and somewhat lower in the ICV/IP group (1.8±1.4) than in the control group (2.2±1.4), but not in the IP group (2.2±1.2) (Fig. 4). Furthermore, morphologic scores significantly correlated with the lipid/NAA and lipid/Cr ratios (Fig. 5).
DISCUSSION
Recently 1H-MRS has been used as a quantitative noninvasive assessment tool in monitoring of brain development and in the diagnosis of neurologically damaged infants (3-5). 1H-MRS can detect metabolites such as NAA and other acetyl compounds, which serve as primarily neuronal markers; Cr, including phosphocreatine and Cr, which are bioenergetic markers; choline-containing compounds (Cho), which are released during membrane disruption; and lactate (Lac), which accumulates in response to anaerobic tissue metabolism (2, 16). Decreased NAA/Cho and NAA/Cr ratio and increased Cho/Cr ratio have were in asphyxiated neonates with poor neurologic outcomes after 1 yr (4, 17). 1H-MRS in asphyxiated neonates has also shown increased lactate and decreased NAA in thalamus, as well as increased Lac and decreased Cr in basal ganglia (18, 19). During hypoxic-ischemic injury, there is a significant increase in the lipid peak, which correlates with apoptotic cell death, as well as in the intensity of the lipid peak, which is directly related to the apoptotic cell count (14). We therefore used the lipid/NAA and lipid/Cr ratios as apoptotic markers.
Newborn infants subjected to transient hypoxic-ischemic injury during birth asphyxia are apparently relatively normal soon after resuscitation but show evidence of delayed cerebral injury hours later, the magnitude of which predicts the severity of later neurodevelopmental impairment (20, 21). The mechanism of delayed injury is unclear, but apoptotic cells are detected in brains of infants who died after birth asphyxia, suggesting that inappropriate activation of the apoptotic pathway accounts, at least in part, for the delayed cell death (6, 8, 21).
Apoptosis was first described as a type of cell death distinct from necrosis, with no swelling or loss of membrane integrity, and no inflammatory response from the host tissue (22). Apoptotic cells undergo a ubiquitous physiologic process that is essential to the development and survival of multicellular organisms. This process takes place during embryologic development, turnover of gastrointestinal epithelium, and the regulation of the immune system. Many pathological events that cause necrosis, including hypoxic-ischemic injury, can also induce apoptosis (2, 4, 6, 23). Necrosis may predominate in more intense ischemic damage, whereas apoptosis may occur during milder ischemic damage and may take time to develop (24). Immature cortical neurons have been shown to be more susceptible to apoptosis than mature neurons, perhaps because cells of younger animals more readily undergo apoptosis than cells of more mature animals (21, 25). Thus, blocking the apoptotic cascade may prolong the therapeutic window after hypoxic-ischemic events, especially in the developing brain (6-8).
The ability of specific therapeutic agents to reduce neuronal damage associated with hypoxic-ischemic injury has been tested in animal models. Caspase inhibitors, which have antiapoptotic activity and are believed to play a key role in the delayed neuronal cell death after hypoxic-ischemic injury (7, 9). Caspases are synthesized in most cells as inactive precursors and are subsequently activated. Caspase-3 is a terminal enzyme in the caspase family that activates an endonuclease (caspase-activated DNAse), resulting in DNA fragmentation (26). Caspase inhibitors, including inhibitors of caspase-3, may prolong the therapeutic window after hypoxic-ischemic injury (7, 27, 28).
Recently, GH administration has been reported to inhibit neuronal death during hypoxic-ischemic injury, and to have a neuroprotective effect in the cerebral cortex, hippocampus, and thalamus (10, 11). Although the mechanism is not completely known, several reports suggest that hypoxic-ischemic injury induces neuronal death by downregulating Bcl-2 protein levels, followed by sequential activation of the caspases, and that GH protects neuronal cells by inhibiting alterations in Bcl-2 protein levels and caspase activities (12, 13).
We found that the lipid/NAA ratio was significantly lower in rats administered GH by the ICV and ICV/IP routes, that lipid/Cr ratio was significantly lower in rats administered GH by the ICV, and that the degree of morphologic changes in the brain was significantly correlated with the lipid/NAA and lipid/Cr ratios. In our results, lipid/NAA ratio and lipid/Cr ratio were not significantly changed by IP administration of GH. Because GH does not usually cross the blood-brain barrier, IP administration of GH may not block cell death. Taken together, these findings suggest that GH exerts neuroprotective effects in cerebral hypoxic-ischemic injury by inhibiting apoptosis, especially in the early stage after insult. Our results also suggest that GH, as a caspase inhibitor, can have therapeutic value in neuroprotective effect of hypoxic-ischemic brain injury.


 XML Download
XML Download