

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Alexander disease (AD) is a fatal and progressive leukodystrophy of the central nervous system. The destruction of white matter in a rostrocaudal gradient is accompanied by the accumulation of abnormal deposits, known as Rosenthal fibers (RFs), in astrocytes, which result in cell death.
The clinical phenotype of AD has been divided into three types based on age at onset, i.e., infantile, juvenile, and adult AD. However, clinically the disease is highly variable among patients in the same disease group. Typical features of the infantile form occur before 2 yr old, and present as progressive macrocephaly, developmental delay, spasticity, and seizures. Seizure is a common clinical symptom in AD, but infantile spasms are a less well-known manifestation. Brain magnetic resonance imaging (MRI) reveals predominantly frontal white matter involvement of low signal intensity on T1-weighted images and high signal intensity on T2-weighted images.
Since Brenner et al. (1) discovered a missense mutation in glial fibrillary acidic protein (GFAP) gene in chromosome 17th in AD, DNA analysis has proven to be a valuable diagnostic modality.
CASE REPORT
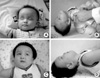
An 8-month-old boy (Fig. 1A, B) was admitted to our emergency room because of repeated seizures. A seizure had occurred a day prior to this presentation. It persisted for 30 min, and his parents observed upward eyeball fixation and tonicclonic movement of the extremities. However, treatment was not sought at the time. At his home on the day of this hospitalization, a seizure redeveloped, persisted for about an hour and then spontaneously stopped. Several hours later the third seizure occurred and did not stop. His parents then brought the patient to our emergency room where the seizure was observed by doctors. Pupils were fixed without deviation with nictitating of eyelids, lip smacking, and stiffness of the right extremities. The seizure stopped after a diazepam injection. Subsequently, seizures were controlled by valproic acid administration.
The patient had no relevant birth or family history. At the time of hospitalization, his body temperature was 36.6℃, pulse rate 128/min, and respiratory rate 28/min. In terms of body measurements, his body weight was 8.8 kg (20th percentile), height 75 cm (90th percentile), and head circumference 51 cm (more than the 97th percentile). Anterior fontanel was soft and flat, and he had macrocephaly and a projecting forehead, similar to that observed in hydrocephalus. Breathing sounds were clear and heart beat regular. Liver and spleen were not palpable and bowel sounds were normal. Light reflex was isocoric and prompt, and deep tendon reflexes were increased. Babinski sign was unremarkable and both ankles showed clonus. He was unable to control his head, and his language and social skills were at the 1-2 month post-natal level.
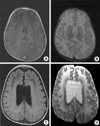
The complete blood count was within normal limits, and total protein/albumin was 6.2/4.3 g/dL, Na/K/Cl 138/4.9/104 mEq/L, blood sugar 100 mg/dL, BUN/Cr 11/0.4 mg/dL, and AST/ALT 26/22 IU/L, respectively. Normal finding was seen by 7.93 (4.9-20.5 mM/M creatinine) in N-acetylaspartic acid (NAA) of urine. Brain MRI showed an absence of myelination except in the frontal lobe and in a portion of the temporal lobe (Fig. 2A, B). DNA analysis revealed that the 716th base sequence of the 4th exon of the GFAP gene showed thymine substituted for guanine (c.716G>T), which led to R239L missense mutation at the 239th amino acid involving the replace of arginine by leucine. Additionally, when we examined the DNA of his parents and older sister, it appeared that they had a normal base sequence (Fig. 3). Interictal waking electroencephalography (EEG) showed slow waves in the frontal lobe, but no epileptic discharge (Fig. 4A).
Three months later, at the age of 11 months, infantile spasms of the flexor type developed accompanied with vocalization after waking up. Interictal sleep EEG showed hypsarrythmia (Fig. 4B). The spasms came under control after starting vigabatrin (40 mg/kg/day) and after the third day of this medication, no more spasm was observed. A follow-up MRI at 24 months of age showed disease progression (Fig. 2C, D).
Now, at age 30 months (Fig. 1C, D), he is in good nutritional condition and has a relatively good cardiopulmonary function. However, he suffers from intermittent vomiting, dysphagia, irritability, easy startling, developmental retardation, slow progressive spasticity of the extremities, and seizures.
DISCUSSION
W. Stewart Alexander, in his original description of a case in 1949 (2), reported a boy with a rapidly enlarging head, hydrocephalus, and developmental delay who died at 15 months of age following an 8-month illness. AD is a progressive and usually fatal neurological disorder in which the destruction of white matter in the brain (3, 4). However, little information is available on the pathogenesis of AD, or on how the GFAP mutation leads to leukodystrophy (5, 6).
In 1976, Russo et al. (7) suggested that AD could be divided into three age-dependent clinical subgroups: infantile, juvenile, and adult and most authors have adopted this classification. However, the clinical picture is highly variable among patients in the same subgroup. The infantile form of AD is most common (8-11), and usually becomes apparent during the first two years of life. Typically patients show both mental and physical developmental delays, followed by loss of milestones, an abnormal increase in head size, and often seizures. The average age of onset is 6 months and the average age of survival after diagnosis is 2 yr and 4 months. But Wakabayashi et al. (12) reported a Japanese case of infantile AD who survived at the age of 25 yr and 7 months. Thus, the clinical progress in AD may be varied by case.
In our patient, although seizure occurred at age 8 months, we suggest that clinical manifestations had developed before at least age 3 months because he was unable to control his head at age 4 months. According to Pridmore et al. (13), seizures often appear at an early stage, whereas spastic quadriparesis is less prominent and occurs later. Macrocephaly is a presenting feature due to rapid head enlargement followed by deceleration. Our patient showed prominent macrocephaly, and developmental retardation before the seizure attacks which occurred at age 8 months. We initially observed slow waves on the frontal area on interictal sleep EEG, but there was no epileptic discharge (Fig. 4A). Three months later, at age 11 months, infantile spasms developed after waking up. An interesting finding of typical hypsarrythmia on interictal sleep EEG (Fig. 4B) was observed. These findings suggested that infantile spasms developed during AD progression. Infantile spasms in AD cases are a remarkable presentation, and are rare according to many reports. Although Gingold et al. (14) described briefly that his AD patient developed multifocal seizures at age 11 months and EEG demonstrated hypsarrhythmia, which was recorded at the left posterior hemisphere during an ictal episode, no further description was provided. They first described an AD case accompanied by infantile spasms, and since no further cases have been published, our case is the second, which is why we consider this case valuable. We controlled these infantile spasms with vigabatrin, which has a highly effective anticonvulsant action.
A definitive diagnosis was only possible by pathological examination of brain tissue, obtained either at biopsy or autopsy, for RFs until the genetic diagnostic method became available. Biochemical studies on compositions of RFs revealed that a large portion of the fibers consisted of the heat shock protein, αB-crystallin (15), and lesser amounts of heat shock protein 27 and ubiquitin (16). Brain MRI is a helpful diagnostic method. In AD, brain white matter degeneration appears remarkably in the frontal lobe on MRI and hydrocephalus occurs infrequently (17, 18).
GFAP was found to be distributed much more on astroglial cell channels comprised of intermediate filaments of diameter 7-11 nm and codes by gene 17q21's GFAP gene. AD can now be diagnosed in a high percentage of cases by analyzing DNA obtained from peripheral blood, which obviates the need for brain biopsy. Brenner et al. (1) sequenced the GFAP coding region in the DNA from 11 unrelated patients who had died of AD and discovered that 10 patients had mutations in the GFAP coding region. Four different GFAP amino acid mutations were observed in this initial group of patients. Li et al. (19) analyzed the DNA of 44 AD cases and found a GFAP gene mutation in 41 cases. Moreover, as studied parental DNAs have also failed to reveal such mutations, it must be presumed that they are wholly spontaneous in nature. This indicates that AD is not autosomal recessive, as had previously been thought by Wohlwill et al. (20) and Schwankhaus et al. (21), but that it is possibility due to a gonadal mosaicism.
In our patient, the R239L GFAP gene mutation (Fig. 3) in which 716th base sequence of 4th exon showed a guanine to thymine substitution (c.716G>T), and as a result, the 239th amino acid of GFAP is changed from arginine to leucine. This was determined by conducting direct base sequence analysis after amplifying all exons and intron regions of the GFAP gene by PCR from peripheral blood genomic DNA. However, the patient's parents and older sister had a normal base sequence. In addition, the position of the GFAP gene mutation in AD which found up-to-date differs from this case. Therefore, the R239L GFAP gene mutation in the present case is presumed to be a de novo mutation or gonadal mosaicism.
In conclusion, the authors report a rare case of AD accompanied by infantile spasms that was diagnosed by GFAP gene analysis in an 8-month-old male with seizures, psychomotor developmental retardation, macrocephaly, and spasticity of the extremities. This DNA analysis performed identified a novel R239L missense mutation.


 XML Download
XML Download