

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Subarachnoid hemorrhage (SAH) produced by a ruptured intracranial aneurysm (IA) affects about 10/100,000 persons annually (1). And, although significant advances have been made in the treatment of aneurismal SAH and vasospasm (2), SAH is currently a significant healthcare problem; moreover, delayed ischemic neurological deterioration (DIND) due to vasospasm remains the greatest cause of morbidity and mortality after SAH (3). The catastrophic consequences of a ruptured IA and the high frequency of IA in the general population demand intensive efforts to define a multifactorial risk model for the identification of those at risk of developing an IA, of rupture of an existing IA, and those at risk of DIND after SAH.
It is known that smoking, alcohol consumption, and hypertension are common risk factors of aneurismal SAH (4). In addition, to these environmental risk factors, genetic factors also play an important role in the pathogenesis of SAH and IAs, as the first degree relatives of patients with the disorder have up to a seven times greater risk than the general population (5), and about 10% of patients with an aneurismal SAH have first or second degree relatives with SAH or unruptured IAs (6). As this familial predisposition is the strongest risk factor for the development of IA (4), the detection of genetic risk factors could provide further diagnostic capabilities.
Disruption of the extracellular matrix is likely to be a factor in the pathophysiology of IA, because IAs are associated with heritable connective tissue and extracellular matrix disorders (7). In this context, the genes involved in maintaining the integrity of the extracellular matrix have been proposed to be the most likely candidates for predisposing an individual to IA development. However, variations in these genes only explain a small proportion of the genetic factors involved in IA (8). Therefore, further studies are needed to identify candidate genes and to help unravel the pathophysiology of this disorder.
Given mounting evidence that genetic factors play a role in IA formation and rupture (1, 9-11), the endothelial nitric oxide synthase gene (eNOS) is being considered as a potential candidate gene that contributes to IA formation and vasospasm after SAH, based on its normal physiologic effects on the cerebral arteries. In the arterial endothelium of animals and humans, the synthesis of nitric oxide (NO) from L-arginine is catalyzed by the enzyme eNOS (12), and the continuously generated NO serves to maintain basal vascular tone and cerebral blood flow (CBF) (13-15). In addition, when one considers that the preferential location of IAs is at the bifurcation of the conductive cerebral arteries in the subarachnoid space (16), it can be hypothesized that hemodynamic change in CBF induced by defective eNOS metabolism may affect the cerebral arterial walls lacking or with disrupted internal elastic lamina, and that this facilitates aneurysm formation (16, 17).
In the present study, the authors aimed to elucidate both whether the eNOS T-786C mutant allele is implicated in susceptibility to SAH, and DIND after SAH, and additionally, to determine whether this mutant allele is differentially expressed in those with a small or large ruptured aneurysm.
MATERIALS AND METHODS
Subjects
A total of 249 human subjects were enrolled in this prospective case-control study. Of 136 consecutive patients (cases) diagnosed with aneurismal SAH based on clinical and radiological findings (including cerebral angiography in all cases), 3 were excluded from analysis because of a previous history of cerebral infarction or myocardial ischemia. During same period, 113 controls were recruited from among out-patient clinic patients with various clinical diagnoses. The control inclusion criteria were as follows; no cerebral vascular abnormality, including aneurysm or thrombosis on CT or MR angiography, no history of a vascular headache or myocardial ischemia, and no definite brain lesion on MR imaging. Demographic and medical history and peripheral blood specimen were obtained from all participants. The maximal diameters of ruptured aneurysms were measured by neuroradiologists and the patients were divided by size into 3 groups in-line with the findings of aneurysm natural history studies (18, 19), i.e., small (≤5 mm), medium (6 to 9 mm), and large (≥10 mm). SAH severity on admission was classified based on CT scan findings according to the grading system described by Fisher (Fisher grades 1 to 4) (20). Clinical SAH severity on admission was scored using the Glasgow Coma Scale (GCS) (21). The outcomes of patients with SAH were classified according to the Glasgow Outcome Scale (GOS) (22); favorable outcome group (GOS 1), good recovery; GOS 2, moderate disability; and an unfavorable outcome group GOS 3, severe disability; GOS 4, persistent vegetative state; GOS 5, death. The study protocol was approved by our institutional review board and all subjects enrolled in the study gave informed consent.
Genetic analysis
Genomic DNA was extracted from peripheral blood lymphocytes using a standard protocol. Polymerase chain reaction (PCR) primers for eNOS T-7896C SNP were designed based on GenBank sequences (accession number: NM_000603); 5'-GCA TGC ACT CTG GCC TGA AGT-3'(forward) and 5'-CAG GAA GCT GCC TTC CAG TGC-3'(reverse). PCR assays were carried out using 1.25 U of AmpliTaq polymerase Gold (Applied Biosystem, NJ, U.S.A.), 100 ng genomic DNA, 2.0 mM of MgCl2, and 10 µM of primer. Amplification conditions were as follows: an initial denaturing cycle at 95℃ for 7 min, followed by 35 amplification cycles (denaturation at 94℃ for 30 sec, annealing at 61℃ for 45 sec, and extension at 72℃ for 1 min), and a final extension step of 72℃ for 5 min. PCR products were electrophoresed in a 1.2% agarose gel, and the amplified genomic DNA fragments were extracted from the gel and purified using a QIAquick® gel extraction kit (Quiagen, Hilden, Germany) according to the manufacturer's instructions. Direct sequencing of both strands was performed using BigDye terminator kits (PE Biosystems, CA, U.S.A.) to determine individual genotypes.
Data analysis
When the clinical characteristics of the study participants were considered, hypertension was defined as blood pressures >140/95 mmHg, and diabetes mellitus as a fasting blood glucose level of >126 mg/dL or a random blood glucose concentration of >200 mg/dL.
Continuous variables were compared using 2-tailed unpaired t-tests. Categorical variables were compared by χ2 analysis with Fisher's exact test. Multiple logistic regression analysis with forward stepwise selection was performed to identify predictors of SAH occurrence, ruptured aneurysm size, SAH outcome, and the occurrence of DIND after SAH. Data were analyzed with SPSS (release 11.5; SPSS, Chicago, IL, U.S.A.) and differences were considered significant for p values of ≤0.05. Conformance with the Hardy-Weinberg equilibrium was tested by comparing the observed and expected genotype frequencies of the controls using the χ2 test.
RESULTS
Compared with controls, SAH cases were significantly younger with a preponderance of non-diabetics and smokers (Table 1). The SAH cases at admission had a mean GCS score of 12.9. More than 90% of patients presented with Fisher grade 3 or more, and the mean diameter of ruptured aneurysms was 6.64±2.74 mm. DIND after SAH occurred in 17 patients (12.8%) and more than 80% patients had a favorable outcome (GOS grade 1 or 2) (Table 2).
Among the controls, the frequency of wild type homozygotes (T/T) was 88.5% and of mutant heterozygotes (T/C), 11.5%. No patient or control harbored the mutant C/C homozygote (Table 1). The frequencies of the eNOS T-786C SNP genotypes concurred with the Hardy-Weinberg equilibrium (p=0.516). No significant difference was observed between cases and controls in the distributions of the eNOS T-786C single nucleotide polymorphism (SNP) genotypes (Table 1). With regard to ruptured aneurysm size and the occurrence of DIND after SAH, no significant differences were found in the distributions of the eNOS T-786C SNP genotypes (Table 3). The mean (±SD) ruptured aneurysm diameter of wild-type homozygotes (T/T; 6.75±2.85 mm) was similar to that of mutant heterozygotes (T/C; 6.19±2.23 mm) (p=0.29, t-test).
Multiple logistic regression analysis after controlling for age and sex showed the eNOS T-786C SNP T/C genotype was independently associated with unfavorable outcome of SAH (GOS grade 3-5) (Exp (β)=4.27, 95% CI 1.131-16.108, p=0.032).
DISCUSSION
Much attention has been focused on the contribution made by eNOS SNPs to the development and progression of various systemic vascular diseases (23-26), because of the role played by NO in the regulations of vascular tone and hemodynamic changes. With regard to the cardiovascular system, it has been reported that high numbers of CA repeats in intron 13 and the G894T SNP in exon 7 of the eNOS gene are associated with the risk of coronary artery disease (24, 27), and that the eNOS T-786C mutation is strongly associated with a history of coronary spasm (23). Of eNOS polymorphisms that have been linked with coronary artery disease, the eNOS T-786C mutation is the only one of which functional role has been elucidated. According to the luciferase reporter gene assays performed by Nakayama et al. (23), this mutation reduces eNOS promoter activity and finally causes a reduction in NO levels. Therefore, an association between the eNOS gene variants and coronary artery disease suggests that the eNOS gene could be associated with aneurismal SAH, in which unique changes in cerebral NO levels have been reported immediately after SAH (28).
Despite the potential link between the eNOS gene and aneurismal SAH, no definite evidence confirming such an association has been found. The eNOS T-786C SNP is one of the most frequently investigated mutations because it is located in the eNOS promoter region as thus potentially affects eNOS expression. In a prospective study involving 141 participants (29), it was suggested that the eNOS T-786C mutant allele does not predict susceptibility to SAH or intracranial aneurysm rupture but rather predicts susceptibility to post-SAH vasospasm. However, in the present study, the authors failed to find any relationship between eNOS T-786C SNP and the susceptibility to SAH or to DIND after SAH as did previous studies (29-31). Given the available evidence and the results of the present study, it is evident that the eNOS T-786C SNP is not a major risk factor for IA formation or SAH. However, whether the eNOS T-786C SNP contributes to vasospasm after SAH remains to be determined because the sample sizes of the vasospasm groups subjected to analysis in the studies above were too small to determine the contribution made by this SNP to vasospasm after SAH. To date, a large blood burden is the only consistently demonstrated risk factor predicting cerebral vasospasm after SAH (32).
According to the stepwise multiple logistic regression analysis performed in the present study, the presence of the eNOS T-786C SNP T/C genotype should be considered as significant risk factors of outcome in cases of SAH. Interestingly, the odds ratio for an unfavorable outcome (GOS grade 3-5) after SAH for individuals with the eNOS T-786C SNP T/C genotype was 4.27 (95% CI 1.13-16.11, p=0.032). This result suggests that the eNOS -786C allele causing a reduction of NO level should be associated with an unfavorable outcome of SAH. However, the exact explanation for the unfavorable outcome of SAH in patients presenting the eNOS -786C allele is out of the scope of the present study. Nevertheless, if the result is confirmed by a larger scale study, the eNOS T-786C SNP T/C genotype could be used as a prognostic marker in individuals with SAH.
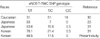
Another controversy concerning aneurysms and the eNOS gene involves the critical size of aneurysm beyond which rupture risk increases appreciably. In the International Study of Unruptured IAs (ISUIA) (19), the rupture rate of aneurysms of ≥10 mm was approximately 20 times that of aneurysms <10 mm among patients without a prior SAH. This result suggests that aneurysm size at the time of diagnosis is an independent predictor of rupture risk. However, the fact that the vast majority of ruptured aneurysms are <10 mm in diameter at the time of diagnosis and treatment (33, 34) reinforces the notion that aneurysm size should not be used as the sole criterion for treatment decision-making. Explanations for this apparent discrepancy include the possibility that there are two distinct subpopulations of IAs; one that develops rapidly and ruptures when <10 mm in diameter and another that enlarges slowly and ruptures when >10 mm in diameter. Recently, Khurana et al. (30) suggested that among Caucasian patients with known IAs that the eNOS T-786C genotype influence aneurismal size at rupture. They found that the mean (±SD) ruptured aneurysm diameter for all heterozygotes (T/C; 8.5±5.2 mm) was significantly greater than that for the C/C (6.0±2.3 mm) or T/T (4.7±1.8 mm) homozygotes (p=0.04). If this finding is reproduced by larger scale study, genotypes could be used to predict the risk of rupture in individuals presenting with unruptured IAs. However, their suggestion is also not free of criticism due to the relatively small size studied (n=52). In addition, according to their suggestion, it should be stated that both the mutant homozygote (C/C) and the wild type homozygote (T/T) influence aneurysm development, which is difficult to rationalize. In the present study of 133 individuals with SAH, no significant differences in the distribution of the eNOS T-786C genotypes were found among the 3 groups defined on the basis of aneurysm size (Table 3). This negative finding is entirely consistent with the result of a recent larger scale multicenter study (n=527) which was performed in East-Asian population that presented with SAH (31). Considering that the distributions of the eNOS T-786C genotypes in normal controls are quite different in these populations (Table 4), discrepancies between Caucasians and East-Asians in terms of eNOS T-786C genotype distributions and their relations with aneurysm size may be attributed to ethnic differences. However, the exact explanation of the ethnic difference remains to be clear.
Taken together, the eNOS T-786C mutation was not found to be associated with either a susceptibility to SAH, or with aneurysm size in Korean population. And the eNOS T-786C SNP T/C genotype could be used as a prognostic marker in individuals with SAH.



 XML Download
XML Download