

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Gentamicin (GM) is an aminoglycoside antibiotic widely used in clinical practice for the treatment of life-threatening Gram-negative infections. However, the clinical use of GM is limited due to its nephrotoxicity (1). GM-induced nephrotoxicity is characterized by direct tubular necrosis, which is localized mainly to the proximal tubule (2). Both in vitro and in vivo studies showed that GM has enhanced the generation of reactive oxygen species (ROS) (2, 3). It is shown that oxidative stress is involved in GM-induced renal damage. GM is shown to enhance the generation of superoxide anion and hydrogen peroxide by renal cortical mitochondria (4). GM treatment induces simultaneous mesangial proliferation and apoptosis in rats.
Heat shock proteins (HSPs), also known as stress proteins and molecular chaperones, play an important role in protecting cellular processes from environmental and physiologic insult, including toxic injury, by preserving the structure of normal proteins and repairing or removing damaged ones (5). A 70-kDa heat shock protein (stress-inducible HSP70, HSP72) has been reported to be a cytoprotective protein in a variety of organs. The induction of HSP is thought to play a protective role in ischemic acute renal failure (ARF). Some findings suggested that pre-induction of HSP72 may prevented lipopolysaccharide-induced liver injury (6). It was also reported that HSP72 could attenuate cisplatin (CDDP)-induced nephrotoxicity. The protective effects of HSP72 are associated with an increased Bcl-2/Bax ratio and decreased apoptosis (7).
Studies demonstrated that prior induction of a heat shock (HS) response protected rats against the lethal effects of sepsis, and HS treatment with subsequent expression of HSP is an effective strategy for tissue protection against damage by endotoxin, various inflammatory mediators, or oxidative injuries (8, 9).
Whether the overexpression of HSP72 can prevent kidney proximal tubular cells from toxic effects of GM is still unclear. In the present study, we induced the overexpression of HSP72 in HK-2 cells, a human proximal tubular cell line, by HS, and observed the protective effects of HS to GM-induced nephrotoxicity.
MATERIALS AND METHODS
Cell culture and treatments
All experiments were performed using HK-2 cells, an immortalized proximal tubular cell line derived from a normal adult human kidney. Cells were cultured in DMEM medium with 10% fetal bovine serum, 100 U/mL penicillin, and 100 µg/mL streptomycin (GiBco BRL Grand Island, NY, U.S.A.). The cultured cells were stored in a humidified incubator (95% air with 5% CO2) at 37℃. These were then separated into four groups which were incubated with medium only, medium containing GM (Sigma Chemicals, St. Louis, MO, U.S.A.) at a concentration of 100 µg/mL, cells were heat shocked at 43℃ for 30 min, and medium containing GM (100 µg/mL) +heat shocked, respectively. MTT assay, lactate dehydrogenase (LDH) release, LDH content, N-acetyl-β-D-glucosaminidase (NAG) and superoxide dismutase (SOD) activities were examined at (24 hr, 48 hr, 72 hr, and 96 hr).
MTT assay
This assay for cell viability is based on the reduction of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, Sisgma Chemicals) by mitochondrial dehydrogenase in viable cells to produce a purple formazan product. At the end of each experiment, cells cultured in 96 well plates (with 100 µL of medium per well) were incubated with MTT (20 µL of 5 µg/mL per well) at 37℃ for 4 hr. The formazan product was solubilized with an addition of 100 µL of dimethyl sulfoxide (DMSO) for 4 hr at 37℃. The dehydrogenase activity was expressed as the absorbance (read with microplate reader) of the formazan product at 570 nm.
LDH release
HK-2 cells were planted into 24-well plates (1×105 cell/mL) and allowed to grow for 1 day. Cell viability after incubation
was estimated by determining the release of LDH from the cells. LDH release was measured by determining LDH activity (measured spectrophotometrically by NADH oxidation at 440 nm) in media.
NAG activity assay
NAG activities were determined using 4-methylumbelliferyl-NAG as substrate, from which NAG liberates 4-methylumbelliferone at pH 4.5. One unit of NAG activity was defined
as the amount of NAG catalyzing the liberation of 1 µM of 4-methylumbelliferone per minute at 37℃. Protein was measured (at 400 nm) by a modified method of Bradford using Bio-Rad Protein Assay (Bio-Rad, Richmond, CA, U.S.A.).
Malondialdehyde (MDA) content assay
Cells were lysed with a cell lysis solution for 30 min at room temperature and the lysate collected. MDA content was determined by the thiobarbituric acid (TBA) assay, as described by Hadley and Draper (10).
SOD activity assay
SOD activity in cell lysate was determined by spectrophotometrically monitoring (at 550 nm) the rate of inhibition of formazan production consecutive to the reduction of 2-(4-iodophenyl)-3-(4-nitrophenol)-5-phenyl tetrazolium chloride (INT) by superoxide anions generated by a xanthine-xanthine oxidase system. Human erythrocyte SOD was used as a standard. One SOD unit was defined as the amount of enzyme that inhibited formazan production by 50% at 37℃.
Immunocytochemistry
Cultured cells were fixed and immunocytochemistry procedure for HSP72 were done as described by Mao et al. (11). Finally, the slides were developed in 0.05% freshly prepared diaminobenzedine solution for 2 min, counterstained with hematoxylin, and then dehydrated, air-dried, and mounted. PBS was used to substitute the primary antibody as a negative control. Photomicrographs were prepared with a Spot 2 digital camera and software (Diagnostic Instruments, Inc., Sterling Heights, MI).
Western blotting analysis
Western blot analysis of the HSP72 level was carried out according to a previously published procedure (10). Briefly, after total protein in HK-2 cells was extracted. Aliquots (10 µg) of proteins were separated by SDS-PAGE on 6% acrylamide Laemmli minigels and transferred onto nitrocellulose membranes overnight. Membranes were blocked for 60 min with 5% nonfat milk in PBS containing 0.5% Tween 20 and incubated for 2 hr at room temperature with HSP72 mouse monoclonal antibody (200-fold dilution) at 4℃ overnight. After incubation with peroxidase-conjugated secondary antibody for 60 min at room temperature, blots were developed using the ECL detection system.
For each sample, the optical density (OD)×area (mm2) was determined using a GS-690 densitometer (Bio-Rad). The HSP content of each sample was calculated from the standard curves of HSP70 or HSP90.
RT-PCR
HSP72 mRNAs were characterized by RT-PCR of total RNA prepared from cultured cells, isolated by TRIzol reagent (Invitrogen, Grand Island, NY, U.S.A.) and quantified by spectrophotometry. The isolated total RNA was amplified by RT-PCR using mRNA selective PCR kit Ver.1.1 (TaKaRa, Japan). After RT reaction at 42℃ for 30 min to synthesize cDNA from mRNA, samples were subjected to PCR for 30 cycles under the following conditions: denaturation at 94℃ for 1 min, annealing at 55℃ for 1 min, extension at 72℃ for 1 min, and a chase at 72℃ for 2 min. The oligonucleotide primer used for RT-PCR and PCR comprised of a forward primer (5'-TTTCGAGAGTGACTCCCGTT) and a reverse primer (5'-AAAGGCCAGTGCTTCATGTC), and for human GAPDH: TCACCACCATGGAGAAGGCT and AAGGCCATGCCAGTGAGCTT. PCR products were visualized on 1% agarose gels stained with ethidium bromide. Control reactions were run using GAPDH 25 cycles of denaturation (94℃ for 30 sec), annealing (58℃ for 30 sec), and extension (72℃ for 1 min) with a final extension of 72℃ for 10 min.
RESULTS
Protective effects of HS from cytotoxicity induced by GM in HK-2 cells
Cytotoxicity of GM in HK-2 cells was assessed by MTT reduction, LDH release, and NAG activities. After exposure to GM at 100 µg/mL for 24 hr, 48 hr, 72 hr, and 96 hr, MTT reduction was observed (Fig. 1). MTT uptake decreased significantly after 24 hr, and then gradually until 96 hr. On the other hand, cells treated with GM+HS did not show reduction at 24 hr. From 48 hr to 96 hr, MTT reduction was highly evident at this group, but less significant than with GM-treated cells. MTT uptake of HK-2 cells treated with HS was higher than that of GM group from 48 hr to 96 hr.
LDH release was used as a marker of cytotoxicity. It increased time-dependently from 24 hr to 72 hr (Fig. 2). Although the increase of LDH release in GM+HS group was observed from 24 hr to 96 hr, it was lower than that of the GM-treated cells. After cells were heat shocked, LDH release increased from 24 hr to 72 hr, but it is lower than that of GM group.
NAG activity was also used for evaluation of nephrotoxicity. This finding is similar to that of the LDH release. NAG activity increased time-dependently from 24 hr to 72 hr (Fig. 3). On the other hand, cells treated with GM+HS did not show significant increase at 24 hr. Although the increase in NAG activity was observed from 48 hr to 96 hr, it was lower than that of GM-treated cells. Results also showed NAG activity of heated shocked cells was higher than cells in control group at 96 hr.
Effects of HS on oxidation in HK-2 cells
MDA levels was evaluated to estimate the degree of lipid peroxidation on cell culture. Fig. 4 showed that GM caused concentration-dependent and time-dependent increases in MDA levels. Meanwhile, HS partially inhibited the increase in MDA content in GM+HS group. SOD activity was used to detect scavenging superoxide anion ability of cells. Total SOD activity diminished in the GM group compared to the control group from 24 hr to 96 hr (Fig. 5). HS slightly prevented the decrease in total SOD activity in GM+HS group. This was similar to that of the MTT reduction assay. Both MDA level and SOD activity of cells which were heat shocked did not show significant change compared with control group.
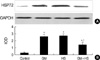
HSP 72 expression after heat shocked and exposed to GM
After cells were heat shocked or exposed to GM at a concentration of 100 µg/mL for 72 hr, the rate of HSP72 positive cells increased dramatically (Fig. 6). It was higher than the control group at 72 hr after cells were exposed to GM+HS, but lower than the cells exposed only to GM. Densitometric measurements of HSP72 bands showed that the expression increased markedly after the exposure to GM or HS for 72 hr (Fig. 7). Proteins extracted from cells exposed to GM+HS showed that the expression of HSP72 increased than that of in control group, but was lower than GM group. Furthermore, from the results of RT-PCR, the change pattera of HSP72 pattern gene transcription showed similar result with the expression pattern of HSP72 (Fig. 8). An increased production of HSP72 gene is associated to a parallel rise of HSP72 protein content.
DISCUSSION
Although GM is widely used as an agent for the treatment of life-threatening Gram-negative infections, its nephrotoxic effect limits its use. Nephrotoxicity induced by GM is a complex phenomenon characterized by an increase in serum creatinine and blood urea nitrogen, and severe proximal renal tubular necrosis followed by deterioration to renal failure (12, 14). It has been clearly shown that oxidative stress is involved in GM-induced renal damage (2, 3); the oxidative damage also has been supported by the fact that the administration of several compounds with antioxidant properties, ROS scavengers, and/or antioxidant enzymes are able to ameliorate the severity of GM-induced renal damage.
In the present study, GM inhibited the proliferation of HK-2 cells, a human kidney tubular cell line, at the concentration of 100 µg/mL time-dependently. LDH release increased from 24 hr to 96 hr after HK-2 cells were treated by GM. Although NAG activity did not increase at 24 hr, it was elevated from 48 hr to 96 hr markedly. Both LDH release and NAG activity are indicatives of cell damage. The increase of them, in the cell culture medium, suggested the injury of cell membrane. Furthermore, GM increased MDA content and decreased SOD activity in vitro. It indicated that GM was effective in inducing oxidative process and inhibiting scavenging O2-.
The highly conserved HSPs accumulate in cells exposed to heat and a variety of other stressful stimuli. HSPs, which function mainly as molecular chaperones, allow cells to adapt to gradual changes in their environment and to survive in other lethal conditions. The HSP70 family (HSP70s) is the most conserved and best studied class of HSPs, which contribute to cell survival after various pathological and physiological stresses. A number of studies demonstrated that HSP70s are inducible in tubular epithelial cells following renal ischemia/reperfusion (15, 16) and toxic injury (17, 18). They play an essential role in protein metabolism under both stress and normal conditions.
HSP72 is one of the members in HSP70 family. Classified as a readily inducible protein, HSP72 can protect a variety of cells, including renal tubular cells, from thermal, toxic, and ischemic injuries in vitro (19). Transfection of LLC-PK1 cells with HSP72 offers protection against cisplatin (CDDP)-induced kidney tubular damage (20) and increased levels of intracellular HSP72 correlate with resistance to CDDP in tumor cell lines (21). Some findings show that administration of geranylgeranylacetone (GGA) to induce the expression of HSP70 could protect vestibular sensory cells from GM ototoxicity, and delays GM-induced vestibular hair cell death (22). It has been demonstrated that GGA ameliorated ischemic acute renal failure via induction of HSP70. Mikami reported that induction of HSP72 could protect cirrhotic rats against lipopolysaccharide-induced liver injury (6). However, whether overexpression of HSP72 could protect kidney proximal tubular cells from GM cytotoxicity has been unknown.
In our present study, HSP72 expression increased parallel with the degree of HK-2 cell injury at 72 hr. The expression of HSP72 was also significantly induced due to the HS at 43℃. However, if cells were treated by GM after HS, HSP72 expression was lower than that of cells exposed only to GM. HSP72 expression was induced markedly after HK-2 cells were seriously damaged by GM. In addition, after HS, the expression of HSP72, LDH release, and NAG activity were less than that of in HK-2 cells treated by GM only. This finding suggested that HSP72, as a group of protective protein, was involved, at least in part, in the GM-induced HK-2 cell injury. Pretreatment with HS attenuated the GM-induced HK-2 cell damage.
Evidence suggests that ROS may be involved in GM-induced ARF, since it has been found that O2-, H2O2, and HO increased with GM treatment (23-25) and it is known that several agents that scavenge or interfere with ROS production successfully ameliorate GM-induced nephropathy (26, 27). After HK-2 cells were heat shocked, SOD activity increased and MDA content decreased compared with that of cells treated by GM only. The finding suggested that overexpression of HSP72 may elevate the oxidant scavenge ability and reduce the production of ROS. The accumulation of HSP72 that was induced by GM might protect cells from oxidative injury.
Here, we demonstrated that GM treatment could induce human kidney tubular cell line (HK-2) damage. The expression of heat shock protein 72 increased when cells were injured by GM. On the other hand, heat shock induced the expression of HSP72. This increased expression may protect cells from the damage induced by GM. Heat pre-conditioning results in induction of heat shock proteins including HSP72 that gives a cytoprotective effect against GM induced nephrotoxicity, by the methods of reduction of tissue oxidation, free radical formation, and disorders of protein folding. Heat pretreatment would be a useful tool in prevention of kidney damage in the clinical setting (28). HSP72 may provide new tools for therapies and drug design that maximize preservation or functional restoration.
It is well established that GM may also affect function of more distal nephron segments. GM treatment caused a selective inhibition of Na, K+-ATPase, and subsequently decreased transport in the thick ascending limb (TAL) of Henle's loop (29, 30). TAL is one of the important segments in active reabsorption of NaCl (31), whether overexpression of HSP72 can protect TAL from GM induced damage is still obscure. That is future work we need to further study.







 XML Download
XML Download