

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Gaucher disease (OMIM 230800, 230900, 231000), the most common lysosomal storage disorder, is caused by a deficiency in the activity of the enzyme glucocerebrosidase (Dglucosyl-N-acylsphingosine glucohydrolase, EC 3.2.1.45; GC). Glucocerebroside or glucosylsphingosine, two substrates of this enzyme, accumulate in the lysosomes of cells of the monocyte/macrophage lineage, particularly in the spleen and liver (1,2). Clinically, patients with Gaucher disease are divided into three major phenotypes: chronic nonneuronopathic (type I); acute neuronopathic (type II); and chronic neuronopathic (type III), based on symptoms of the nervous system, the severity of these symptoms, and the age of disease onset (3). The characteristics of patients with neuronopathic Gaucher disease include oculomotor abnormalities, hypertonia of the neck muscles with extreme arching of the neck, bulbar signs, limb rigidity, seizures and occasional choreoathetoid movements, and neuronal loss (1). However, the mechanisms leading to the neurological symptoms of this disorder remain unknown.
In a previous study, we analyzed the mRNA expression profile in the brain of a mouse model for Gaucher disease using DNA microarrays and found that apoptosis was associated with neuronal loss (4). Moreover, proinflammatory cytokines were upregulated in the Gaucher mouse brain. TNF-α and IL-6 mRNA expression levels in the brains of Gaucher mice were 2.5-fold and 8.5-fold higher than that of wild-type mice, respectively. This implies that inflammation is associated with neuronal degeneration in Gaucher disease. There are some reports of elevated proinflammatory cytokines and sustained inflammatory reactions in the liver of Gaucher patients and the Gaucher mouse (5-7). Inflammation has also been reported in the brains of mouse models for lysosomal storage diseases. In mouse models of Niemann-Pick type C disease, Tay-Sachs disease, and Sandhoff disease, activation of microglia and astrocytes, and secretion of proinflammatory cytokines, such as TNF-α and interleukin 1α(IL-1α), are increased (8,9). Moreover, deletion of the gene for macrophage-inflammatory protein 1α retards neurodegeneration and extends the lifespan in a mouse model for Sandhoff disease (10).
In this study, we investigated the levels of proinflammatory cytokines and their possible role in neurodegeneration in Gaucher mice.
MATERIALS AND METHODS
Mouse care
Glucocerebrosidase-deficient mice (C57BL/6J-Gbatm1Nsb, 'Gaucher mice') were obtained from the Jackson Laboratory (Bar Harbor, ME, U.S.A.). This mouse, which is homozygous for the GC gene-knockout, expresses <4% of normal GC activity, stores glucocerebroside in the lysosomes of cells of the reticuloendothelial system, and dies within a day of birth (11). All the animals were housed in the Korean Food and Drug Administration, which is accredited by the American Association for the Accreditation of Laboratory Animal Care, and were treated according to the Korean Food and Drug Administration and National Institutes of Health guidelines for animal care.
Cell culture
Microglia were prepared from the cerebral cortices of postnatal ICR-strain mice, and from the brains of wild-type mice and Gaucher mice (12). Brain tissues, devoid of meninges and blood vessels, were dissociated by a mild mechanical trituration. The isolated cells (1×106) were seeded in 75 cm2 T flasks in MEM medium (Life Technologies, Grand Island, NY, U.S.A.) containing 10% heat-inactivated fetal bovine serum, for two weeks. Microglia were then detached from the flasks by mild shaking and seeded in plates with or without 200 µM conduritol B epoxide (CBE; Sigma, St Louis, MO, U.S.A.), a specific GC inhibitor(13), at 37℃ in a 5% CO2 incubator for 8 days.
Semiquantitative reverse transcriptase-polymerase chain reaction (RT-PCR)
Fetal brains of postnatal wild-type mice and Gaucher mice were separated into cerebral cortex, brainstem, and cerebellar regions. Total RNA was isolated using an RNeasy Mini kit (Qiagen GmbH, Germany). Reverse transcriptase (RT) reactions were performed with 2 µg of total RNA using 50 µg/mL of oligo (dT) primer at 42℃ for 1 hr in the presence of 20 units of MMLV RT (Promega, Madison, WI, U.S.A.). Aliquots of the RT product were amplified by polymerase chain reaction (PCR) using 20 cycles of 94℃ for 45 sec, 60℃ for 30 sec, and 72℃ for 30 sec. The PCR cycles were performed in a reaction volume of 25 µL, including 2 µL RT products as a template, 1×reaction buffer, 1 unit of Ex Taq DNA polymerase (TaKaRa, Japan), and 20 pmol of each primer. Primer sequences were as follows: β-actin, forward 5'-CCCACACTGTGCCCATCTAC-3', reverse 5'-AGTACTTGCGCTCAGGAGGA-3'; IL 1α, forward 5'-CGTCAGGCAGAAGTTTGTCA-3', reverse 5'-GTGCACCCGACTTTGTTCTT-3'; IL 1β, forward 5'-CAGGCAGGCAGTATCACTCA-3', reverse 5'-AGGCCACAGGTATTTTGTCG-3'; IL 6, forward 5'-GTTCTCTGGGAAATCGTGGA-3', reverse 5'-GGAAATTGGGGTAGGAAGGA-3'; and TNF-α, forward 5'-ACGGCATGGATCTCAAAGAC-3', reverse 5'-CGGACTCCGCAAAGTCTAAG-3'. The PCR products were analyzed on 1% agarose gels.
Detection of cytokines
To measure cytokines secreted from microglial cells, we used the Beadlyte Mouse Multi-Cytokine Detection System 2 (Upstate, Lake Placid, NY, U.S.A.) following the manufacturer's instructions. Fifty microliter aliquots of media from cultured microglia were loaded into 96-well microplates with 25 µL of Luminex bead-conjugated anti-mouse multi-cytokine antibody. Two hours later, 25 µL of biotin-conjugated antimouse multi-cytokine antibody was added to each well. After incubation for 1.5 hr, 25 µL of streptavidin-phycoerythrin solution was added. After 30 min, 25 µL of stop solution was added. Samples were analyzed using a Luminex 100 luminometer (Luminex, Austin, TX, U.S.A.).
Determination of nitric oxide and reactive oxygen species
The amount of nitric oxide (NO) produced from microglia of wild-type mice and Gaucher mice was determined using the Griess method (14). Using 96-well cell culture plates, each well was filled with 100 µL of MEM medium mixed with an equal volume of Griess reagent (0.1% naphthylethylenediamine, 1% sulfanylamide, 2.5% H3PO4). After 10 min at room temperature, the optical density of samples was measured using a microplate reader at 550 nm. To measure intracellular reactive oxygen species (ROS) production, cells isolated from wild-type and Gaucher mouse brains were loaded with 10 µM 5,6-chloromethyl-2', 7'-dichlorodihydrofluorescein diacetate acetyl ester (CM-H2DCFDA, Molecular Probes, Eugene, OR, U.S.A.) for 30 min at room temperature in the dark. The cells were then washed, resuspended in phosphate buffered saline and analyzed on a flow cytometer (FACS Vantage, BD, Franklin Lakes, NJ, U.S.A.) equipped with a 488 nm argon-ion laser and a 525 nm bandpass emission filter.
RESULTS
Genes for the cytokines IL-1α, IL-1β, IL-6, and TNF-α were upregulated in the cerebral cortex, brainstem, and cerebellum of Gaucher mice compared with those of wild-type mice by RT-PCR (Fig. 1A). We induced Gaucher disease in a cell model using primary cell cultures of microglia from ICR-strain mice. After incubation with 200 µM CBE for 8 days, IL-6 and TNF-α mRNAs were upregulated in CBEtreated microglia (Fig. 1B). However, there was no increase in IL-1α or IL-1β mRNA levels (data not shown). Using the Beadlyte Mouse Multi-Cytokine Detection System 2, which can detect 10 cytokines (MG-CSF, IFN-γ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12 and TNF-α), we analyzed cytokine levels in CBE-treated microglia. Three of them were detectable and secreted at high levels from CBE-treated microglia (Fig. 1C). The mean level of secreted IFN-γ was 2.2-fold greater in CBE-treated microglia (35.3±5.3 pg/mL) compared with control microglia (16.2±5.9 pg/mL). The amount of secreted IL-6 was 1.8-fold higher in CBE-treated microglia (219.5±49 pg/mL) compared with control microglia (125.8±9.9 pg/mL), and the level of TNF-α secreted from CBE-treated microglia (183.8±25.2 pg/mL) was 2.8-fold higher compared with control microglia (65.3±4.1 pg/mL).
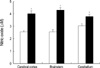
Cells from the cerebral cortex, brainstem, and cerebellum of Gaucher mice produced 4.04±0.19 µM NO, 4.31±0.18 µM NO, and 3.78±0.20 µM NO, respectively. The same tissues from wild-type mice produced 2.57±0.09 µM NO, 2.58±0.15 µM NO, and 3.03±0.17 µM NO, respectively (Fig. 2). Thus, the level of production of NO in cells of Gaucher mice was 1.25-fold higher in the cerebellum (p<0.01) and 1.67-fold higher in the brainstem (p<0.001) compared with NO production in the corresponding cells of wild-type mice.
ROS generation in microglial cells of the mouse brain was analyzed using flow cytometry (Fig. 3). Microglial cells from the brainstem and cerebellum of Gaucher mice had higher levels of ROS compared with microglial cells from the brainstem and cerebellum wild-type mice at embryonic day 19.5 (E19.5) and postnatal day 1 (P1). However, there was no difference in ROS levels in cells from the cerebral cortex of wild-type mice and Gaucher mice.
DISCUSSION
Gaucher disease is mainly caused by the accumulation of glucocerebroside, a metabolic intermediate in the synthesis and degradation of complex glycosphingolipids. This causes the release of calcium from intracellular stores, leading to enhanced sensitivity to agents that induce neuronal death (13,15). The deacylated analog of glucocerebroside, glucosylsphingosine, accumulates in the cerebral and cerebellar cortices of patients with type II and type III Gaucher disease (16), and in the brains of Gaucher mice (17), and has been suggested to be a candidate neurotoxin (18,19). However, no explanation has yet been proposed to link these experimental results to the known neurodegenerative symptoms of Gaucher disease (20).
Gaucher disease is characterized by the accumulation of glucosylceramide in macrophages and macrophage-derived cells in various tissues (3). Among the earliest and more provocative suggestions to explain phenotypic variability in Gaucher disease was the suggestion that it is an immune or autoimmune reaction to a chronic storage of glucocerebroside (21). Systemically, an excessive immune response involves the inhibition of proinflammatory cytokines, such as TNF-α, which is produced by macrophages and is necessary for adhesion molecule expression and leukocyte recruitment to inflammatory sites (22).
Inflammation is an important contributor to neuronal damage in other neurodegenerative conditions, such as Parkinson's disease, Alzheimer's disease, multiple sclerosis, and amyotrophic lateral sclerosis (23-25). One of the best-known features of inflammation in the central nervous system (CNS) is the production of NO, which is a product of macrophages that have been activated by cytokines and a microbial compound found in the inflammatory reaction (26). Lipopolysaccharide, IFN-γ, TNF-α, and IL-1 are responsible for microglial activation and NO production (27). Production of NO is associated with the generation of ROS, including superoxide anions, hydroxyl radicals, lipid hydroperoxides, and hydrogen peroxide. These compounds are toxic to neurons because they induce lipid peroxidation, DNA fragmentation, and protein oxidation (28,29). Moreover, ROS can activate protein kinase C, mitogen-activated protein kinase, and nuclear factor-kappa B, which regulate the expression of genes encoding a variety of proinflammatory factors (30,31). Inflammation-mediated neurodegeneration has received considerable attention in lysosomal storage diseases. In mouse models of Sandhoff disease and Tay-Sachs disease, genes associated with the activation of microglia and astrocytes are overexpressed, and progressive CNS inflammation correlates with the severity of disease (32,33).
We could not observe the inflammatory reaction in the fetal brains of Gaucher mice because this mouse model does not survive longer than one day after birth (11). Therefore, we examined the fetal brains of Gaucher mice and explored the accumulation of glucosylceramide in microglia, which could be a causative factor of inflammation related to neurodegeneration.
In this study, the secretion of proinflammatory cytokines increased in the brains of Gaucher mice and in CBE-treated microglia, a cell model of Gaucher disease. We examined whether the upregulation of proinflammatory cytokines resulted in the production of NO and found increased production of NO in the brain cells of Gaucher mice. Increased ROS generation in microglial cells from the brainstem and cerebellum of Gaucher mice also occurred. Thus, a deficiency of GC activity may mediate microglial activation resulting in the production of cytokines, NO, and ROS.
Further in vivo approaches to understanding the pathophysiology of Gaucher disease and the development of new therapeutic strategies for Gaucher disease have been hindered by the lack of a viable animal model (11,34). However, the generation of viable mouse models of Gaucher disease has recently been reported (35), and these mouse models will be useful for elucidating the pathogenesis of the neuronopathic forms of Gaucher disease.
Although we have no direct evidence that the overexpression of proinflammatory cytokines or the production of NO and ROS are involved in the neurodegenerative symptoms of Gaucher disease, further investigation of the relationship between microglial activation and the accumulation of GC substrates should clarify the roles of these factors.


 XML Download
XML Download