

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cystic fibrosis (CF; MIM 219700) is a lethal disease, which is induced by mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene on chromosome 7q31. CF is one of the most frequently-occurring autosomal recessive disorders in Caucasians, with an incidence of 1 in every 2,000 to 3,000 (1). However, CF is quite rare in the Asian population that an epidemiological study in the Japanese population revealed the incidence of CF being about 1 in 350,000 (2,3). CF is also extremely rare in the Korean population and there have been only a few reports of this condition thus far (4-6). Here, we report a Korean patient with CF, who was diagnosed by both a sweat chloride test and genetic analysis of the CFTR gene.
CASE REPORT
A 15-yr-old boy was admitted to the Samsung Medical Center, complaining of chronic productive cough and dyspnea without any gastrointestinal symptoms. During the first decades of life, the patient had been treated for several episodes of bronchiolitis and pneumonia, necessitating frequent hospital admission. Six years prior to the current hospitalization, the patient's respiratory symptoms became aggravated. He was admitted to a civil hospital for the treatment of pulmonary tuberculosis, and received antituberculosis drug therapy for 12 months. He developed frequent rhinorrhea and nasal stuffiness, and underwent sinus surgery at the age of twelve. His respiratory symptoms progressively worsened, and he was diagnosed with bronchiectasis three years ago. He had a 25-yr-old brother and a 24-yr-old sister, with an unremarkable past and a current medical history.
On admission, the patient's blood pressure was 128/69 mmHg, heart rate 80 beats per minute, respiratory rate 20 breaths per minute, body temperature 36.0℃, height 174 cm, and he weighed 53.6 kg. He was also determined to exhibit 96 percent oxygen saturation when breathing room air. Upon physical examination, diffuse crackles and rhonchi were heard throughout the patient's lungs, with intermittent wheezes.
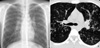
Chest radiographs and computed tomographic (CT) scans of the chest, performed without intravenous contrast material administration, revealed bilateral multifocal bronchiectasis and multiple centrilobular nodules (Fig. 1). Pulmonary function studies showed a forced vital capacity of 2.62 L (63% of predicted), with a forced expiratory volume of 1.36 L per second (36% of predicted). The patient's total lung capacity was measured to be 6.39 L (121% of predicted). A CT scan of his paranasal sinuses evidenced severe inflammatory changes, associated with pansinusitis. Staphylococcus aureus was cultured from his sputum. Mycobacterial and fungal cultures were negative. A tuberculin skin test was also negative. Conditions such as immunodeficiency and ciliary dyskinesia were ruled out by the normal results of serum immunoglobulin tests, and by the finding of normal bronchial mucosal biopsy. There was no evidence of exocrine pancreatic insufficiency and malaborption.
The sweat chloride concentration was measured by a quantitative pilocarpine iontophoresis sweat test, in accordance with the guidelines of the National Committee for Clinical Laboratory Standards (7). The average sweat chloride concentrations on both forearms were 63.0 mM/L, a finding that exceeded the reference limit (<40 mM/L).
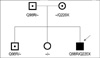
Direct sequencing analysis of all coding exons and their flanking intronic sequences demonstrated that the patient possessed two mutations in his CFTR gene (Fig. 2): a heterozygous A to G transition in exon 4 (c.293A>G; Q98R) and a heterozygous C to T transition in exon 6a (c.658C>T; Q220X). A family study demonstrated that the patient's father and elder brother both also possessed the Q98R mutation, whereas his mother harbored the Q220X mutation. The patient's sister, however, harbored no such mutations. Based on our search of the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr/), Q98R and Q220X mutations have been reported in CF patients from Southern France and Southern England (8,9).
DISCUSSION
CF is the most frequently encountered life-limiting autosomal recessive genetic disorder in Caucasians, with an incidence of 1 in every 3,200 newborns in the United States (1). However, it is less common in African Americans (1 in 15,000) (10) and quite rare in Asians (1 in 350,000 in the Japanese population) (2,3). CF is normally suspected upon the finding of one or more typical CF phenotypical features, including chronic sinopulmonary disease, gastrointestinal/nutritional abnormalities, salt loss syndrome, and obstructive azoospermia in males (7).
CF is caused by mutations in the CFTR gene, which is located on the long arm of chromosome 7. This gene consists of 27 exons, and encodes for an integral membrane protein composed of 1,480 amino acids. Functionally, CFTR regulates the expression of the chloride (Cl-) channel, which is expressed in the apical membrane of exocrine epithelial cells (1). Currently, approximately 1,366 mutations and polymorphisms are listed in the Cystic Fibrosis Mutation Database (http://www.genet.sickkids.on.ca/cftr/). However, only 11 mutations have been reported in the Korean population thus far (Table 1). In the present study, we report two additional mutations in the list of CFTR gene mutations (Q98R and Q220X), which have been identified in Koreans. It is of note that these two mutations have previously been reported in CF patients from Southern France (8) and Southern England (9), respectively, but are not frequently encountered mutations even in the Caucasian population.
As shown in the Table, no mutation except IVS8 T5 variant has repeatedly been observed in Korean population. Furthermore, only two mutations (R117H and delF508) are found among 25 mutations in the screening panel for Caucasian CF patients recommended by the American College of Medical Genetics (11). Therefore, as demonstrated by Lee et al. (12), a possible common mutation might not exist in the Korean population.
Although CF tends to be discovered early in life, the diagnosis of CF is being made in adults with increased frequency (13,14). Patients diagnosed as adults normally present with chronic respiratory problems. As a group, they exhibit a milder variant of lung disease, a lesser degree of Pseudomonas infection, and are more likely to be pancreatic-sufficient than are patients in whom CF is diagnosed at earlier ages (13,14). Physicians who are unfamiliar with the spectrum of CF phenotypic manifestations may not consider CF as a component of their differential diagnoses, thus delaying accurate diagnosis.
In summary, we report here a Korean CF patient, who was diagnosed via a sweat chloride test, as well as a molecular genetic analysis of the patient's CFTR gene. Although CF continues to be quite a rare condition in Koreans, CF should still be suspected in patients exhibiting recurrent and unexplained chronic respiratory symptoms.

 XML Download
XML Download