

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Aspirin (ASA) and non-steroidal anti-inflammatory drugs (NSAIDs) cause bronchoconstriction in 10-20% of adult asthmatics (1). ASA-induced bronchoconstriction is more common in female patients with non-atopic asthma (57% vs. 43% in North America, 73% vs. 27% in Korea) and is associated with acute or chronic rhinosinusitis and/or nasal polyps in 60-80% of patients (2,3). Seventy-nine percent of these patients require long-term oral or inhaled corticosteroid therapy for symptom control (4).
The mechanism of ASA-induced bronchoconstriction is not yet fully understood. It is widely accepted that ASA and NSAIDs block the cyclooxygenase pathway, which causes arachidonate substrates to be diverted to the 5-lipoxygenase pathway (5). A recent review has emphasized the importance of chronic overproduction of cysteinyl leukotriene (5). Downregulation of cyclooxygenase-2 and prostaglandin E2 may also accelerate cysteinyl leukotriene production (5,6).
On the other hand, organ- and non-organ-specific autoantibodies have been described in asthmatics by several investigators (7-9). It has also been suggested that non-atopic patients have an increased incidence of autoantibodies and a higher tendency to have accompanying autoimmune diseases, which include rheumatoid arthritis, Sjögren's syndrome, and autoimmune thyroiditis (7). Furthermore, our previous study (12) demonstrated that circulating IgG to bronchial epithelial cells, which was identified as being directed against cytokeratin (CK)18, was present in the sera of patients with non-atopic asthma. Although antinuclear antibody (ANA) and Clq-binding immune complex (CIC) have been evaluated previously in ASA-intolerant asthma (9), there have been no reports on the quantification of levels of autoantibodies against bronchial epithelial cytokeratins. Recent studies have demonstrated that human leukocyte antigen (HLA) DPB1*0301, which is generally accepted as a contributor to genetic susceptibility in various autoimmune diseases, is a valuable gene marker for ASA-intolerant asthma in both Caucasian and Oriental populations (10,11).
In this study, to investigate the involvement of an autoimmune mechanism in ASA-intolerant asthma, we compared the levels of circulating autoantibodies, which included ANA and autoantibodies to tissue transglutaminase (TGase), CK8, CK18, and CK19, in the sera of 79 patients with ASA-intolerant asthma to those in the sera of patients with ASA-tolerant asthma and healthy control subjects. In addition, circulating serum CIC was detected by enzyme-linked immunosorbent assay (ELISA).
MATERIALS AND METHODS
Subjects
Seventy-nine patients with ASA-intolerant asthma (30 men and 49 women: mean age of 46 yr), and two sex-matched control groups: 61 patients with ASA-intolerant asthma (20 men and 41 women; mean age of 43 yr), and 88 normal healthy controls (33 men and 55 women; mean age 36 yr), who were enrolled at the Department of Allergy and Rheumatology, Ajou University Hospital, Suwon, Korea.
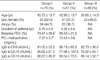
Asthma was diagnosed according to the revised GINA guidelines (2002), and comprised typical asthma symptoms, reversible airway obstruction, and airway hyperresponsiveness to methacholine. ASA-intolerant asthma was diagnosed by positive results in the lysine-aspirin (L-ASA) bronchoprovocation test (BPT). A patient was classified as having allergic asthma if a skin prick test with at least one of fifty common aeroallergens was positive and correlated with the clinical symptoms. In patients with non-atopic asthma, both skin prick tests and serum-specific IgE were negative. All of the ASA-tolerant patients showed negative responses on L-ASA BPT and had no history of adverse reactions to ASA and NSAIDs. The 65 healthy controls were enrolled at random from a local population. All of the subjects gave informed consent for the studies, and the protocols were approved by the ethics committees of Ajou University Hospital, Suwon, Korea. The demographic data on the three study groups are compared in Table 1.
Allergy skin prick test
Skin prick tests were performed with 50 common aeroallergens (Bencard, Bretford, U.K.), which included Dermatophagoides pteronyssinus, D. farine, cat, dog, cockroach, tree pollen mixture, grass pollen mixture, mugwort, ragweed, Humulus japonicus, Aspergillus, Alternaria, histamine, and a saline control.The reactions were read after 15 min, and the wheal was measured in two directions. A positive reaction was defined as a mean wheal diameter of ≥3 mm. Atopy was determined by a positive skin test response to at least one common inhalant allergen.
Bronchoprovocation test with methacholine and L-ASA
The methacholine bronchoprovocation test was performed according to the method previously described (10). Five inhalations of normal saline at 5-min intervals were taken followed by a series of successively doubled doses of methacholine (0.075-25 mg/mL) until a 20% fall in FEV1 was observed or the maximum dose given. FEV1 was measured 5 min after the beginning of each set of inhalations of aerosolized methacholine. The methacholine PC20 level was determined by interpolation from the dose-response curve.
The L-ASA BPT was performed according to the modified method described previously (10). All medications, including xanthine derivatives, beta-2 agonists, and steroids, were stopped 72 hr before the procedure. The test solutions were delivered by a Devilbiss 646 nebulizer (DeVilbiss, Somerset, PA, U.S.A.) connected to a compressed air source (5 L/min). L-ASA powder (Althargyl, DongAh, Korea) that contained 900 mg L-ASA with 100 mg aminoacetic acid was diluted in normal saline, to produce a stock solution of 300 mg/mL. The stock solution was diluted in normal saline to produce a range of doubling concentrations from 75 to 300 mg/mL. A placebo solution of normal saline was used as the control. Subjects inhaled the aerosol via a mouthpiece during normal tidal breathing. If the baseline forced expiratory volume in 1 sec (FEV1) was >65% of the predicted value, and the change in FEV1 was <10% after inhalation of normal saline, L-ASA BPT was performed. The L-ASA solution was inhaled every 30 min. The FEV1 and maximum mid-expiratory flow were measured at 10 and 30 min after each dose. The provocation was stopped when the FEV1 had decreased >20% from the baseline. When the change in FEV1 at 30 min after inhalation of the maximum dose of L-ASA was <15%, the last dose was inhaled once more. The FEV1 and maximum mid-expiratory flow (MMEF) after the last dose were measured at 10, 30, and 60 min, and then hourly for 7 hr.
ELISA for specific IgG antibodies to CK8, CK18, and CK19
The serum-specific IgG level was detected by ELISA using 50 µL of diluted (1:20) patient serum or negative control serum. This was determined to be the optimal concentration in the preliminary experiment, and was added to each well of ELISA plates that had been coated with 100 ng/well of CK8, CK18, and CK19 (Research Diagnostics, Flanders, NJ, U.S.A.) dissolved in PBS. The wells were blocked with 200 µL of blocking buffer (PBS that contained 10% FBS). After 1 hr of incubation at room temperature, the plate was washed three times with PBS-T. Alkaline phosphatase-conjugated anti-human IgG (100 µL; Sigma, St Louis, MO, U.S.A.), which was diluted to 1:10,000 v/v with 10% FBS-PBS, was added to the wells for 1 hr at 37℃. The wells were then washed with PBS-T, and PNPP (p-nitrophenyl phosphate; Sigma) was added as the substrate solution. The reactions were stopped by the addition of 1 N NaOH. The optical density of the solution was determined at 405 nm using an ELISA reader. The final absorbance value was determined by subtraction of the absorbance value of the uncoated well. Each absorbance value is presented in arbitrary units based on the standard curve, which was derived from serial dilutions of the pooled serum samples with high specific levels of IgG to CK8, CK18, and CK19, respectively. The positive cutoff value was determined as the mean plus two standard deviations (SD) of the absorbance values of the serum samples from 89 unexposed healthy control subjects.
Measurements of ANA, IgG and IgA antibodies to TGase, and CIC levels
Serum levels of ANA, IgG and IgA antibodies to TGase, and CIC were measured by ELISA using commercially available kits (BL-Diagnostika GmbH, Mainz, Germany), according to the manufacturer's instructions.
IgG immunoblotting for CK8, CK18, and CK19
Recombinant CKs in a solution of 30 mM TrisHCl (pH 8.0), 2 mM EDTA, 9.5 M urea, 2 mM DTT, and 10 mM methylammonium chloride were separated by discontinuous SDS-polyacrylamide gel electrophoresis (PAGE) using a 4% to 20% Tris-glycine gradient gel (Invitrogen, Carlsbad, CA, U.S.A.). After electrophoresis, the proteins were transferred onto a polyvinylidene difluoride membrane (Millipore, Bedford, MA, U.S.A.) and blocked with 5% non-fat milk. After the transfer, the membrane strips were probed with monoclonal antibodies to CK8, CK18, and CK19 (Sigma) at 1:20 dilutions for 3 hr at room temperature. After washing, the strips were incubated with alkaline phosphatase-conjugated goat anti-human IgG (Sigma) for 1 hr at room temperature. After a final wash, the membranes were stained with the substrate solution nitro-blue tetrazolium/5-bromo-4-chloro-3-indoyl phosphate (Sigma); the intensities of the stained bands were measured using a densitometer and presented as relative intensities.
Statistical analysis
The results are expressed as mean±standard deviation. The Student's t-test and analysis of variance (ANOVA) with a post-hoc test (Dunnett's) were used for comparisons between continuous data such as age, levels of autoantibodies and CIC, duration of asthma, baseline FEV1, and PC20 methacholine dose. Intergroup comparisons of the prevalences of specific IgG antibodies to the three CKs, ANA, IgG antibodies to TGase, CIC, and clinical parameters were done with the chi square and Fisher's exact tests, with adjustments for multiple comparisons. For association study between presence of autoantibodies and the disease-related phenotypes, we accepted a p-value using Levene's test for equality of variances in order to exclude an artificial false significance. All of the described tests were two-tailed. A p value ≤0.05 was regarded as statistically significant. The statistical analyses were performed using the SPSS 11.0 for Windows software (SPSS Inc., Chicago, IL, U.S.A.)
RESULTS
Clinical characteristics of the study subjects
In Group I, 34 (45.3%) subjects had atopic tendencies and 53 (67.6%) had chronic rhinosinusitis. The mean baseline FEV1 value was significantly lower for Group I than for Group II (p=0.001). However, the prevalence of atopy and duration of asthma, as well as the PC20 methacholine doses, showed no significant differences between the two asthma groups (Table 1).
Levels of IgG specific for CK8, CK18, and CK19
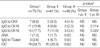
There were no significant differences in the levels of IgG to CK8 and CK18 among the three groups, while the levels of IgG to CK19 differed significantly between Groups I and III (Dunnett's t-test, p=0.024). The prevalence of IgG to CK19 (17.7%) was highest in Group I, followed by those of IgG to CK18 (13.9%) and IgG to CK8 (8.9%). However, statistical significance was noted only for the prevalences of IgG to CK18 and of IgG to CK19, compared between Groups I and III (p=0.014, p=0.020, respectively). IgG antibodies to CK19 were significantly associated with IgG to CK8 (Pearson's coefficient=0.320, p<0.001) and CK18 (Pearson's coefficient=0.442, p<0.001).
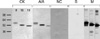
IgG immunoblotting for CK8, CK18, and CK19
Fig. 1 shows the SDS-PAGE and IgG immunoblot findings using patient sera, buffer control, negative control sera, and monoclonal antibodies to CK8, CK18, and CK19. Specific binding to CK8, CK18, and CK19 was noted in the sera of ASA-intolerant asthma subjects.
Association between Anti-CK antibodies and asthma phenotypes and the prevalences of other autoantibodies
Table 2 summarizes the relationships between the different autoantibodies detected in the study subjects. Three (3.8%) subjects in Group I and four (6.8%) subjects in Group II had detectable IgG antibodies to TGase, while none of the subjects in Group III had IgG antibodies to TGase. None of the study subjects had IgA to TGase. One subject of Groups I (1.3%) and II (1.7%), respectively had high serum levels of ANA, while none of the subjects in Group III had serum ANA. The prevalence of CIC was significantly higher in Groups I (24.7%) and II (33.3%) than in Group III (0%). However, there were no significant associations between anti-CK antibodies and IgG to TGase, ANA, and CIC (p>0.05 for all) in Group I. In addition, no significant associations were found between the prevalence of IgG to the three CKs and the presence of HLA DPB1*0301 (p>0.05 for all; data not shown). Regarding asthma phenotypes, the presence of anti-CK18 and anti-CK19 antibodies was significantly associated with PC20 methacholine values in the analysis with both asthma groups (p=0.001 and p=0.003, respectively, Table 3). However, no significant difference was noted between anti-CKs antibodies and PC20 methacholine, baseline FEV1 in the analysis with subjects limited to Group I.
DISCUSSION
In this study, we have demonstrated that serum anti-CK8, anti-CK18, and anti-CK19 autoantibodies are present in certain populations of bronchial asthma patients. The prevalences of anti-CK18 and anti-CK19 autoantibodies were significantly higher in patients with ASA-intolerant asthma than in healthy controls. While some of the ASA-tolerant asthmatics had anti-CK18 and anti-CK19 antibodies, the prevalences did not differ from those of normal controls. Furthermore, a proportion of the asthma patients, regardless of ASA sensitivity, had laboratory markers of autoimmunity, including ANA, CIC, and IgG antibody to TGase, although the prevalences of these markers were too low to be statistically significant.
The mechanism of induction of autoantibodies in asthma remains unknown. Disruption of bronchial epithelial cells and subsequent exposure of autoantigens or ineffective antigen elimination during the inflammatory process may cause chronic immune stimulation and autoantibody production. In asthma, the bronchial epithelium is characteristically damaged, with shedding of the columnar cells into the airway lumen. Recently, it has been demonstrated that high doses of acetaminophen reduce the levels of glutathione in lung tissues (13), and that the asthmatic bronchial epithelium is more susceptible to oxidant-induced apoptosis (14). During this early apoptosis, activated caspases cleave a variety of structural proteins. Therefore, it may be postulated that disruption of the cytoskeleton leads to the loss of apoptotic cells from the epithelium and that the altered epithelium becomes an important source of autacoid mediators, chemokines, and growth factors, which contribute to ongoing inflammation (15,16).
Cytokeratin is a cytoskeletal structure that is expressed only in epithelial cells. Pairs of keratins seem to be consistently co-expressed in different types of epithelial cells. Thus, CK8, CK18, and CK19, which were used in this study, have been found only in simple epithelia, including both bronchial and lung alveolar epithelial cells (17), which are the major target tissues of asthma. Previously, CK18 has been identified as a bronchial epithelial autoantigen that is associated with non-allergic asthma (12). In isocyanate-induced asthma, CK18 has been identified as a major diisocyanate-binding protein (18), and significantly higher levels of serum IgG to CK19 have been detected (19). CK8 and CK18 contain the caspase cleavage site and have been reported to undergo marked re-organization during apoptosis (16). These findings raise the possibility that fragments of CKs and intracytoplasmic materials are released to the blood vessels and may play a role in the formation of circulating autoantibodies, including ANA and IgG to CKs and TGase.
Recent in vitro studies have indicated that the opsonization of extracellular keratin aggregates by IgG-anti-CK autoantibodies plays an important role in promoting the phagocytosis of cytokeratin aggregates (21). This may relate to our results, which show that asthma patients with anti-CK18 and anti-CK19 antibodies have higher prevalences of CIC as well as more severe airway hyperresponsiveness to methacholine. These results suggest that persistent airway inflammation in some patients with bronchial asthma results from a non-IgE-mediated reaction to endogenous or exogenous antigen, possibly an autoantigen, or to a chronic viral infection (22). These possibilities are supported by a number of studies, which have shown that some patients with ASA-intolerant asthma have elevated markers of autoimmunity with rheumatic symptoms. Enhanced IgG4 synthesis in association with viral infection and a positive association with the HLA gene marker have been noted (10,11). Our previous study has demonstrated that HLA-DPB1*0301 is a valuable gene marker for ASA-intolerant asthma (10). However, in the present study, no direct relationship was found between this HLA gene marker and the prevalences of IgG antibodies to the three CKs, ANA, IgG antibody to TGase, and CIC. Moreover, none of subjects with these autoantibodies complained of rheumatic symptoms.
Tissue transglutaminase (TGase I) is a member of the Ca2+-dependent enzymes that catalyze the cross-linking of proteins. TGase I is expressed in tissues that contain simple epithelia, such as bronchial epithelia, skin epidermis, liver, gastrointestinal tract, kidney, and endothelial cells (23). The induction and activation of TGase is part of the apoptotic cascade and plays an effector role in the stabilization of apoptotic bodies to limit the leakage of intracellular components into the extracellular space (24). Recently, anti-TGase antibodies have been found not only in various autoimmune diseases, such as celiac disease, diabetes mellitus type I, and SLE, but also in toluene diisocyanate-induced asthma (19). Based on these findings, we examined the impact of anti-TGase antibodies on persistent airway inflammation in patients with ASA-intolerant asthma, to evaluate the potential of TGase as an endogenous antigen. However, the prevalence of IgG to TGase in patients with ASA-intolerant asthma was <5%, which was similar to that in ASA-tolerant patients, and it had no effect on asthma phenotype.
Considering the results of the present study, an autoantibody-mediated mechanism for ASA-intolerant asthma appears to be unlikely. We cannot rule out the possibility that the existence of anti-CK and anti-TGase autoantibodies is a non-specific consequence of epithelial injury, since anti-CK18 and anti-CK19 antibodies have been reported in patients with various airway diseases, which included non-atopic (13) or TDI-induced asthma (19), pulmonary fibrosis (25), adenocarcinoma (26), and non-airway diseases like an autoimmune hepatitis (27). Besides, the positivity levels of these autoantibodies were neither high enough nor related to ASA-sensitivity, atopy, asthma duration or baseline FEV1. However, long-term prospective studies are needed to address whether autoantibodies cause clinical symptoms of autoimmunity several years later, have a pathogenic role, or merely reflect the epiphenomenon of polyclonal immune stimulation.
A mechanism by which IgG antibody to CK could be involved in the pathogenicity of asthma has been suggested in the case of hexamethylene diisocyanate-induced asthma. Inhaled hexamethylene diisocyanate can bind to the CK of epithelial cells, which may induce T cell and cytokine production (18). Considering the clinical features of the patients with ASA-intolerant asthma enrolled in our study, with most of the patients having moderate to severe persistent asthma, we hypothesize that self-antigen or possibly CK-driven immune responses contribute to the development of persistent airway inflammation in these patients. Patients who are more susceptible to epithelial damage following airway inflammation, or who have impaired clearance of apoptotic bodies, may develop serum IgG antibodies to CK. It is clear from this study that asthmatics with circulating anti-CK18 and anti-CK19 autoantibodies suffer from more severe airway hyperresponsiveness and have more complement-binding immute complexes.
The prevalence of IgG to CK18 in non-atopic asthma was a little lower than that seen in the previous investigation using immunoblot analysis (13). This difference may be due to the different antigen and antibody detection methods used; the ELISA system using recombinant CK was applied in this study to screen a larger population of patients with ASA-intolerant asthma.
In conclusion, we have confirmed the existence of several autoantibody types, with the highest prevalence of IgG to CK18, IgG to CK19, and CIC in patients with bronchial asthma, irrespective of ASA sensitivity or atopy. In addition, the correlation with airway hyperresponsiveness suggests that anti-CK18 and anti-CK19 autoantibodies are involved in the persistent airway inflammation of bronchial asthma. To confirm the role of anti-CK autoantibodies, further studies are needed to show the presence of autoreactive helper T cells specific for CKs and the deposition of activated complement components in patients with bronchial asthma.

 XML Download
XML Download