

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pfeiffer syndrome (PS) is a rare autosomal dominant congenital disorder, originally described by Pfeiffer in 1964, and is characterized by an acrocephalic skull, regressed midface, syndactyly of hands and feet, and broad thumbs and big toes, with a wide range of variable severity (1-3). PS is known to be caused by mutations in exons IIIa or IIIc of the fibroblast growth factor receptor (FGFR) 2 or FGFR 1 gene (4-7). We report with a review of literature the first neonate in Korea with PS who had bicoronal craniosynostosis, bilateral syndactyly of the fingers and toes, and broad thumbs and big toes.
CASE REPORT
A girl was delivered by spontaneous normal vaginal delivery at 38 weeks gestation after an unremarkable pregnancy. She was 3.5 kg in weight (25-50 percentile), the head circumference was 33 cm (25-50 percentile), and the height at birth was 52 cm (25-50 percentile). Her mother is a gravid 4, para 3, 40 yr-old Korean woman. Her parents are both phenotypically normal and nonconsanguineous. The patient is the 2nd among 3 children and is the only affected child.
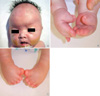
Upon birth, the infant demonstrated bilaterally fused coronal sutures, mild proptosis, and maxillary hypoplasia (Fig. 1A). The soft tissues of her 2nd, 3rd and 4th fingers were fused bilaterally to the proximal portion of distal phalanx and her bilateral thumbs were broad and deviated radially (Fig. 1B). Syndactyly of the feet involving all 5 toes was observed (Fig. 1C). The infant demonstrated evidence of respiratory distress with choanal hypoplasia. There was no deformity or motion limitation of bilateral elbows or shoulder joints.
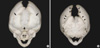
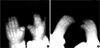
Computed tomogram (CT) and magnetic resonance imaging (MRI) studies of the cranium revealed no other abnormalities except for the presence of craniosynostosis. Three-dimensional reconstruction CT showed 3-dimensional virtual images of the bilaterally fused coronal sutures (Fig. 2A) but that the sagittal and lambdoid suture were open more than normal to compensate for the small intracranial space, and resulting overall in a brachycephalic head shape (Fig. 2B). Plain radiography of her hands and feet showed no bony fusion between the phalanges (Fig. 3). Three-dimensional CT image of choanal air airway showed small and narrow airway of choanal space (Fig. 4). She did not have any visceral abnormality upon ultrasonographic examination of the abdomen and heart.
The patient had a decompressive endoscopic tunnel craniectomy at 4 weeks of age, and is planned for second surgical correction of craniosynostosis, followed by additional correction procedure for the finger and craniofacial anomalies at a later date.
DISCUSSION
The exact incidence of PS is unknown, but is expected to be 1 in every 100,000 births in the Western population, and approximately 60 cases to date have been reported in the past world-wide literature (8, 9). However, it is rarer in Asian population: only several cases were reported in Japan (10, 11) and no reports as yet in Korea. Cohen (12) in 1993 classified this syndrome into 3 clinical subtypes and suggested that these subtypes might not be classified as separate entities, even though these classifications have important diagnostic and prognostic implications. The classic PS is designated type 1. Type 2 consists of a cloverleaf skull with Pfeiffer hands and feet, together with ankylosis of the elbows. Type 3 is similar to type 2 without the cloverleaf skull. In type 3 PS, ocular proptosis is severe, and the anterior cranial base is markedly short. Various visceral malformations have been found in association with type 3 (12). Early demise is characteristic of both types 2 and 3, which to date has been reported to occur only as sporadic cases (7, 9). Clinical manifestations of this patient appeared to fit into subtype 1 according to Cohen's clinical classification of PS.
The etiology for this syndrome is autosomal dominant or fresh mutations in type I conditions, and sporadic in types II and III. PS is genetically heterogeneous. Some cases are linked to mutations at the FGFR 1 gene at chromosome 8p11.22-p12 (4). Mutations at the FGFR2 gene, which map at chromosome 10q25-q26, have also been reported recently (9). These mutations are related to the old age of parents, especially the father, because these mutations are suspected to confer a selective advantage to survival of sperm (13). Usually, the parents are not affected and the risk for future children of that couple is minimal. The offspring of a patient with PS have a 50-60% chance of inheriting the syndrome due to the dominant characteristics of this gene.
PS may involve craniosynostosis that is most often of the coronal and lambdoid, and occasionally sagittal sutures. A severe form of craniosynostosis in the PS is Kleebattschädel (cloverleaf anomaly). The abnormal skull shape could result in a small intracranial volume that increases intracranial pressure, which might be commonly presented as headaches or visual changes. Some children with PS may have only one or both mid-face problems and craniosynostosis. The most common features of the face are regressed mid-face and shallow orbits, which may be present at birth but sometimes become more evident as the childhood progresses. Underdeveloped maxillary bone of the face results in very shallow orbits and exophthalmos or proptosis. The shallow orbits and proptosis may lead to damage of the cornea from dryness, or exposure keratosis. Maxillary hypoplasia also results in a small larynx and pharynx behind the nose and mouth. This restricts the passage of air into the trachea and lungs and causes respiratory distress, particularly at night when snoring and snuffling can interrupt sleep. Similarly, the passage of food is restricted, and regurgitation may result in aspiration of food into the lungs. Stone et al. (14) in 1990 described an infant with PS in whom the trachea showed replacement of the cartilaginous rings by a solid cartilaginous plate extending the full length of the trachea and beyond the carina resulting in tracheal stenosis.
Some other features commonly seen in these patients are visual disturbances related to an imbalance of the muscle that move the eyes and hearing loss due to recurrent ear infections. Vallino-Napoli (15) in 1996 reviewed the audiologic and otologic features of 9 patients with PS, ranging in age from 2 to 12 yr. Hearing loss was found in 8 of the 9 patients. The degree of hearing loss varied but was moderate to severe in most patients. Seven patients had conductive hearing loss and 1 had mixed loss; none had purely sensorineural loss. Primary CT findings were stenosis and/or atresia of the external auditory canal, hypoplasia of the middle ear cavity, and an enlarged middle ear cavity. The arrangement of the teeth, or dentition, is also affected. Rarely, there may be palate problems. Moore et al. (3) reported upper airway compromise in three of four patients with PS type 2, and five of seven patients with PS type 3, and cited this as the cause of death in two cases. McCarthy et al. (16) found upper airway anomalies in 5 of 15 patients with PS of unspecified severity, and these were the primary cause of death in two patients. Congenital anomalies of the upper airway are clearly an important cause of morbidity and mortality in PS type 2 and 3 but are rare in PS type 1 (14).
The hands and feet in PS are involved to variable degree (17-19). The thumbs and big toes are broad and deviated radially. Patient may have mild soft tissue webbing of syndactyly between the second, third, and fourth digits of either or both hands and feet. The digits may be short and misshapen. Whilst the fusion of the fingers and toes in Apert syndrome always involves fusion of the soft tissues of the second, third and fourth digits, this manifestation is similar with PS, but often there is fusion of the bones as well as soft tissues in Apert syndrome (17).
Other anomalies in PS could include a cleft palate, choanal atresia, tracheo- and broncho-malacia, cloverleaf skull, fused vertebrae, Arnold-Chiari malformation, hydrocephalus, and imperforate anus. After birth, seizures and mental retardation may develop.
The above case presented by authors is in accordance with type 1 PS because the infant had a craniosynostosis only of bilateral coronal suture and demonstrated broad and radially deviated bilateral thumbs and big toes that are characteristic of PS, even though she also had syndactyly of the bilateral 2nd, 3rd and 4th fingers only with soft tissue webbing. The diagnosis of the Apert syndrome is appropriate rather than PS if a patient has broad thumbs and syndactyly with bony fusion (17) or syndactyly of the second, third and fourth web spaces (19). However, clinical differential diagnosis has been confusing until now in relation with genetic findings because Pfeiffer mutation has been reported to occur in a patient with Apert syndrome (20), and identical mutations in the FGFR2 gene could cause both PS and Crouzon syndrome phenotypes (21) as illustrated by the demonstration of both phenotypes of Apert and of PS in experimental mice (22). Some authors have attempted to revise the classification for Apert, Carpenter, Crouzon, Jackson-Weiss, Pfeiffer and Saethre-Chotzen syndromes on a genetic basis. Because of widely intermingling of phenotypic expressions or genetic sharing of the same mutation, as yet there is no definite evidence that phenotypic expressions correlate accurately with genetic mutations (10, 20-24). This is probably because the gene that controls these expressions has not been elucidated to date (25).
The prognosis depends on the severity of associated anomalies, mainly the severity of the central nervous system compromise. Patients with type 1 syndrome have, in general, a good prognosis. Patients with types 2 and 3 usually expire early in infant or childhood even though some may survive with aggressive medical and surgical management (5).
Multiple staged surgeries are the general treatment plan for patients with PS. In the first year of life it is preferred to release the synostotic sutures of the skull to allow adequate cranial volume and thus allowing for brain growth and expansion. In this patient, we performed a decompressive endoscopic craniectomy at 4 weeks of age for decompression of the high intracranial pressure due to coronal synostosis and maxillary hypoplasia. Secondary decompressive skull remodeling is planned at 4-6 months of age, and additional skull expansion surgery would be repeated as the child grows to about 2 or 3 yr of age. Although significant malformation of the fingers and toes are present, usually these function adequately and do not require the surgical correction. However, this patient had soft tissue webbing between the fingers and toes, and thus will require surgical dividing procedure after infancy for future active physical therapy. Mid-facial advancement may be performed in order to provide an adequate orbital volume and reduce the exophthalmos, and to correct the dental occlusion to achieve appropriate functional position, but these procedures are not common in patients with PS. This patient also had severe choanal hypoplasia with respiratory distress from maxillary hypoplasia that will be corrected with mid-facial advancement at 5-6 yr of age.

 XML Download
XML Download