

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Inflammation is a dynamic response of vascularized tissues to injury and an important part of the body's defense mechanisms (1). Although inflammation is importantly required for homeostasis, excessive inflammation may induce various diseases including cancer (2-5). Cigarette smoking has been known to induce cancer in a variety of tissues including lung through chronic inflammation (6). Previous studies have shown that cigarette smoking induces proteins related in inflammation, so called inflammation markers (7, 8). Tumor necrosis factor alpha (TNF-α) and white blood cells (WBCs) are well known inflammatory markers. Cytokines such as TNF-α are important inducers of inflammatory response, and the existing evidence implicates the role of TNF-α in inflammatory pathways that increase a variety of diseases (9-13). In one large prospective study (Honolulu Heart Program), persons with WBC count in highest tertile had 30% increased risk of dying from cancer compared with persons in the lowest tertile (14). Another prospective study has shown that elevation in WBC count within the clinically normal range is associated with increased risk of several chronic diseases related to inflammation (15). Therefore, it is believed that increased TNF-α level and WBC count play important roles in smoking-induced inflammation which could be associated with increased risk of diseases (16).
Glutathione S-transferase (GST) M1 and P1 are detoxification enzymes, which have a pivotal role in catalyzing the conjugation of glutathione (GSH) to electrophilic substrates (17, 18). Because of this function of GSTM1 and GSTP1, they may protect against inflammation and oxidative stress through detoxification of various toxic substances in tobacco smoke (19, 20). Several studies have demonstrated that individuals lacking the GSTM1 activity have increased risk of smoking-related diseases (21, 22). Moreover, the GSTP1 protein was reported to be very important in metabolism of many carcinogenic compounds including benzo[a]pyrene diolepoxide (BPDE) which is one of the most important carcinogenic metabolites in tobacco smoke (20).
The human GSTM1 is polymorphic and its deletion is responsible for the existence of a null type lacking the enzyme function (23). Furthermore, a polymorphic site at codon 105 (A to G substitution replacing isoleucine with valine) of GSTP1 is known to change the kinetic property of the enzyme in binding with a few of electrophilic substrates including PAH diolepoxides (24). For this reason, GSTM1 and GSTP1 gene polymorphisms may contribute to susceptibility to inflammation-related diseases. These results also suggest that GSTM1 and GSTP1 gene polymorphisms may be involved in the detoxification of toxic substances in cigarette smoke to change the levels of TNF-α and WBC, which are inflammatory markers. Therefore, in the present study we tried to investigate whether tobacco smoke exposure increases the level of inflammation biomarkers and the GSTM1 and GSTP1 gene polymorphisms are associated with inflammatory response due to tobacco smoke exposure.
MATERIALS AND METHODS
Study population and sampling
A total of 300 students at Inha University, Incheon, Korea, were recruited during September 2002 after exclusion of students having conditions affecting inflammatory biomarkers such as inflammatory disorder or medication of anti-inflammatory drugs. Information on demographic characteristics and lifestyle habits including smoking and alcohol consumption were collected by trained interviewers using a structured questionnaire. We controlled fasting state by asking them for overnight fasting to participate in this study and samplings of blood and urine were done in the morning. After collecting venous blood samples from subjects, whole blood in a heparinized tube was kept at 4℃ for counting of blood WBC and serum in a plain tube was separated by centrifugation within 4 hr and frozen at -70℃ for the analysis of TNF-α level. In addition, urine was kept frozen at -20℃ for the analysis of cotinine level. The study protocol was approved by the institutional review board at Inha University Hospital and written informed consent was provided by all study subjects.
Measurement of serum TNF-α levels and blood WBC counts
Serum TNF-α level was determined by enzyme-linked immunosorbent assay (ELISA) according to the manufacturer's instructions (R&D Systems, Minneapolis, MN, U.S.A.). The detection limit of the assay was 0.12 pg/mL. In addition, the count of blood WBC was measured using cell counter.
Measurement of urinary cotinine levels
Urinary cotinine levels were determined for monitoring tobacco exposure. After urine samples were centrifuged to remove particulate matter, cotinine level was analyzed by a liquid phase radioimmunoassay method using the fundamental principles of a competitive reaction between antibodies and 125I-labeled cotinine and urinary cotinine (DPC, Los Angeles, CA, U.S.A.). After allowing antigen-antibody reaction for 30 min, the fraction bound to antibody was precipitated and its radioactivity was measured for 1 min using Cobra D5010 Quantum, a gamma-counter (Packard, Meriden, CT, U.S.A.). Urinary cotinine level was adjusted to urine creatinine.
Genotyping assays
Genomic DNA was extracted from peripheral blood lymphocytes using a QIAamp DNA Blood Mini Kit (Qiagen, Valencia, CA, U.S.A.), and analyzed using PCR and RFLP method for GSTM1 and GSTP1 gene polymorphisms. The 20 µL PCR reaction mixture used for GSTM1 and GSTP1 genotyping contained 10 mM of Tris-HCl (pH 9.0), 40 mM of KCl, 1.5 mM of MgCl2, 0.25 mM of each dNTP, 1 unit of Taq polymerase (Bioneer, Seoul, Korea), 20 pmole of the forward and reverse primers, and 50 ng of genomic DNA as a template. Amplifications were performed using the following conditions; initial denaturation at 94℃ for 5 min, 35 cycles with denaturation at 94℃ for 1 min, annealing at 65℃ for 1 min, and extension at 72℃ for 1 min, and then completion at 72℃ for 7 min. PCR amplification of reaction mixtures was carried out using a thermal cycler, PTC-200 (MJ Research, Watertown, MA, U.S.A.).
GSTM1 and GSTP1 genotypes were determined using a multiplex PCR method. Primer sets for GSTM1 and GSTP1 genes were used for the amplification reaction. The primers designed were: GSTM1-forward, 5'-GAACTCCCTGAAAAGCTAAAGC-3', GSTM1-reverse, 5'-GTTGGGCTCAAATATACGGTGG-3'; GSTP1-forward, 5'-CCAGTGACTGTGTGTTGATC-3', GSTP1-reverse, 5'-CAACCCTGGTGCAGATGCTC-3'. The PCR products were digested with restriction enzyme BsmA I at 55℃ for 3 hr for GSTP1 genotyping and detected by electrophoresis on 3% 3:1 NuSieve/agarose gel (Cambrex Bio Science, Rockland, ME, U.S.A.). Genotyping of the GSTM1 gene was determined based on the presence of a 215-bp product. For the genotyping of the GSTP1 gene, the 189-bp PCR product remained intact for the A allele, but was cleaved into smaller fragments of 149-bp and 40-bp in the case of the G allele. For confirming the analyses, ten percent of the samples were randomly chosen and genotyped again, producing identical results.
Statistical analysis
Means and the distributions of characteristic variables according to smoking status were compared using the t-test or chi-square test. The t-test was used to analyze relationships between TNF-α level or WBC count and subject characteristics such as age, sex, body mass index (BMI), smoking status, cotinine level, and the GSTM1 and GSTP1 genotypes. The effects of smoking on total serum TNF-α level and blood WBC count were evaluated by linear regression analysis. Correlation between smoking amounts obtained from questionnaire and urinary cotinine level was evaluated by Spearman's and Kendall's rank correlations as well as Pearson's correlation analysis. Analysis of variance (ANOVA) was used to evaluate the effects of genotype combinations on inflammatory markers according to smoking amount or cotinine level. For smokers, pack year value was calculated to indicate cumulative cigarette dose (pack years=[cigarettes/day÷20]×years of smoking). A probability level of 0.05 was used as the criterion of statistical significance based on two-sided test. SAS, version 8.1 (SAS institute Inc., Cary, NC, U.S.A.) was used
for the statistical analysis.
RESULTS
The demographic characteristics of the study subjects by smoking status are listed in Table 1. Mean pack year of smokers was 3.56 (range, 0.15-9.00). BMI and the GSTM1 genotype showed no significant differences with respect to smoking status. However, urinary cotinine level, age, sex, alcohol consumption, and the GSTP1 genotype were found to be significantly different in distribution according to smoking status (cotinine, p<0.01; age, p<0.01; sex, p<0.01; alcohol consumption, p<0.01; and GSTP1, p=0.02, respectively).
Total serum TNF-α levels and blood WBC counts according to age, sex, BMI, alcohol consumption, and GSTM1 and GSTP1 genotypes are listed in Table 2. Age, sex, and alcohol consumption were not found to be significantly related to TNF-α level or WBC count. Obesity was found to be significantly associated with increase of WBC count (p<0.01), but the observed increase of TNF-α with obesity did not reach statistical significance (p=0.08). Subjects with the GSTM1 null genotype had significantly higher TNF-α level than those with the GSTM1 present genotype (p=0.04). However, the GSTM1 genotype did not affect WBC count (p=0.82). Subjects with the GSTP1 AG or GG genotype had significantly higher WBC count than those with GSTP1 AA genotype (p=0.04). TNF-α level was also higher in subjects with the GSTP1 AG or GG genotype than those with the GSTP1 AA genotype, although this difference did not reach statistical significance (p=0.38).
Cotinine level has been used to differentiate active smokers from nonsmokers because cotinine is widely considered to be the best marker for monitoring tobacco exposure in either actively or passively exposed individuals. However, as shown in Table 3, the information of smoking amount obtained from questionnaire showed better relationships with inflammatory biomarkers (TNF-α, p=0.18; and WBC, p<0.01, respectively) than urinary cotinine level (TNF-α, p=0.37; and WBC, p=0.04, respectively) after adjustment of age, sex, and BMI, although smoking amount and cotinine level showed good correlation (Pearson, correlation coefficient=0.56, p<0.01; Spearman, correlation coefficient=0.83, p<0.01; and Kendall, correlation coefficient=0.70, p<0.01, respectively).
The effects of smoking on serum TNF-α level and blood WBC count were evaluated after stratification according to the GSTM1 or GSTP1 gene polymorphism to estimate whether the GSTM1 and GSTP1 gene polymorphisms modify the relation between smoking and serum TNF-α level or blood WBC count. In the linear regression analysis after adjustment of age, sex, and BMI, none of the GSTM1 and GSTP1 genotypes showed statistically significant effect on the relation between serum TNF-α level or blood WBC count and smoking exposure measured by smoking amount or urinary cotinine level (all of four strata, p>0.1). Since the metabolism of the toxins from tobacco smoke is extremely complex, single gene may not have enough impact on inflammation. In an attempt to address this complexity, we constructed and analyzed composite genotypes (Fig. 1, 2). Combination of GSTM1 null and GSTP1 AG or GG genotypes in subjects with pack year or cotinine level over zero showed higher TNF-α level than the other genotype combinations, although the differences were not statistically significant (ANOVA, pack year, p=0.07; and cotinine, p=0.15, respectively) (Fig. 1). This trend was also shown in linear regression (pack year, ptrend=0.07; and cotinine, ptrend=0.15, respectively). However, combination of GSTM1 null and GSTP1 AG or GG genotypes in subjects with pack year or cotinine level over zero did not show higher WBC count than the other genotype combinations, although subjects with pack year or cotinine level over zero showed increased WBC count than those with pack year or cotinine level zero in all genotype combinations (ANOVA, pack year, p=0.34; and cotinine, p=0.15, respectively) (Fig. 2).
DISCUSSION
TNF-α protein has been known to play an important role in a variety of diseases through inflammatory reaction and tobacco smoke has been known to induce inflammation-related diseases including cancer (6-11). Therefore, inflammationrelated diseases caused by smoking may be promoted through increase of TNF-α level. However, the effect of smoking on TNF-α level was inconsistent in previous reports. Kuschner and coworkers reported that smokers had higher level of pulmonary TNF-α than nonsmokers (6), but Keatings and coworkers did not show the elevated TNF-α level in asymptomatic smokers (25). Our data also did not support the significant association between smoking amount and TNF-α level. However, when we analyzed the effect of smoking on TNF-α level according to genotype combination, smoking showed marginally significant effect on TNF-α level in subjects with combination of GSTM1 null and GSTP1 AG or GG genotypes in spite of low level of tobacco smoke exposure. Moreover, WBCs were significantly increased by smoking regardless of genotypes. These results indicate that smoking can increase the levels of inflammation biomarkers, particularly in subjects with combination of GSTM1 null and GSTP1 AG or GG genotypes. The relationship between smoking and inflammation-induced carcinogenesis is biologically plausible. Oxidative stress such as the formation of reactive oxygen species (ROS) is a key component of inflammation, and they lead to damage of macromolecules such as DNA, protein, and lipid, eventually cancer when ROS are generated in large amounts (19, 20, 26). Moreover, GST enzymes have been reported to protect against endogenous or exogenous oxidative damage (27, 28). These facts support that GST enzymes may protect against inflammation through oxidative damage induced by toxic chemicals contained in tobacco smoke. Although host antioxidant defences such as GSTs detoxify ROS, individuals differ in their ability to deal with oxidant burden, and such differences are, in part, genetically determined (29). Inability to detoxify ROS due to genetic variation is likely to perpetuate the inflammatory process which could be associated with a variety of diseases.
We hypothesized that elevated TNF-α is a good indicator of inflammation caused by smoking and serum TNF-α level would be associated with GST polymorphisms in relation to smoking. The present study showed that subjects with the combination of GSTM1 null and GSTP1 AG or GG genotypes have higher TNF-α level than those with the other genotype combinations. This result suggests that there are interactions between GSTM1 and GSTP1 genetic polymorphisms, thus subjects with the combination of GSTM1 null and GSTP1 AG or GG genotypes are more susceptible to smoking-induced inflammation than those with the other genotype combinations. The previous study by Ryberg and coworkers support the interaction between GSTM1 and GSTP1 genetic polymorphisms (30). In their study, when the effects of combined GSTM1 and GSTP1 genotypes on DNA adduct formation were examined, lung cancer patients with GSTM1 null and GSTP1 AG or GG genotype combinations had significantly higher carcinogen-derived DNA adduct level in lung tissue than those with the other genotype combinations. This genotype combination effect was not found in WBC count in our study. The reason may be that the elevation of WBC count may be relatively late and consequential event rather than causal event in inflammatory pathway while TNF-α is considered as a proinflammatory cytokine and a causal factor of inflammation because the binding of TNF-α to its receptors results in the activation of major transcription factors such as AP-1 and NFkB which in turn induce genes involved in acute and chronic inflammatory responses (9). Moreover, TNF-α may be induced through excessive oxidative stress by toxic chemicals contained in tobacco smoke although the level may be modified by GSTM1 and GSTP1 enzymes. For this reason, TNF-α may be more relevant as a biomarker indicating inflammation which could be associated with a variety of diseases than WBC count, although both TNF-α and WBC are indicators of inflammation.
In our study, questionnaire information of smoking showed better relationship with TNF-α level or WBC count than urinary cotinine level, although cotinine is widely considered to be the best marker for monitoring tobacco exposure. Study subjects sometimes intend to stop smoking before study and half life of cotinine in the body is very short (about 17 hr). Therefore, change of smoking habit before study may lead to the change of urinary cotinine level, which may undermine relationship between urinary cotinine level and TNF-α level or WBC count. In addition, our study showed borderline significance of smoking on TNF-α level, probably due to small sample size. Therefore, this study results should be confirmed through studies of larger subjects. In conclusion, our study findings suggest that smoking may induce inflammation measured as TNF-α level or WBC count and combinations of the GSTM1 and GSTP1 polymorphisms may modify the effect of smoking on serum TNF-α level, indicating genetic susceptibility to smoking-related diseases.
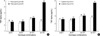




 XML Download
XML Download